I. A. Kovács, A.-L. Barabási
Destruction perfected
Nature (News & Views) 524, 38-39 (2015)
Read the abstract
Pinpointing the nodes whose removal most effectively disrupts a network has become a lot easier with the development of an efficient algorithm. Potential applications might include cybersecurity and disease control. See Letter p.65, by F. Morone and H. A. Makse (Supplementary 1).
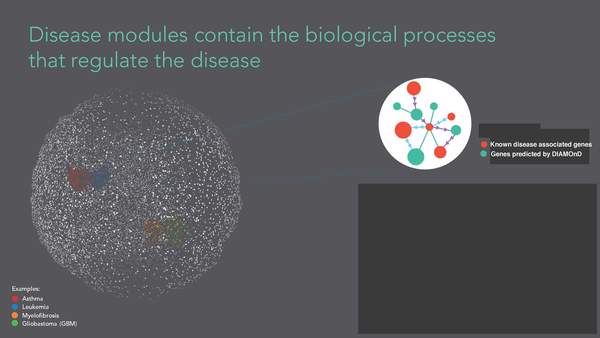
S. D. Ghiassian, J. Menche, A.-L. Barabási
A DIseAse MOdule Detection (DIAMOnD) Algorithm Derived from a Systematic Analysis of Connectivity Patterns of Disease Proteins in the Human Interactome
PLOS Computational Biology pcbi.1004120, 1-21 (2015)
Read the abstract
The observation that disease associated proteins often interact with each other has fueled the development of network-based approaches to elucidate the molecular mechanisms of human disease. Such approaches build on the assumption that protein interaction networks can be viewed as maps in which diseases can be identified with localized perturbation within a certain neighborhood. The identification of these neighborhoods, or disease modules, is therefore a prerequisite of a detailed investigation of a particular pathophenotype. While numerous heuristic methods exist that successfully pinpoint disease associated modules, the basic underlying connectivity patterns remain largely unexplored. In this work we aim to fill this gap by analyzing the network properties of a comprehensive corpus of 70 complex diseases. We find that disease associated proteins do not reside within locally dense communities and instead identify connectivity significance as the most predictive quantity. This quantity inspires the design of a novel Disease Module Detection (DIAMOnD) algorithm to identify the full disease module around a set of known disease proteins. We study the performance of the algorithm using well-controlled synthetic data and systematically validate the identified neighborhoods for a large corpus of diseases.
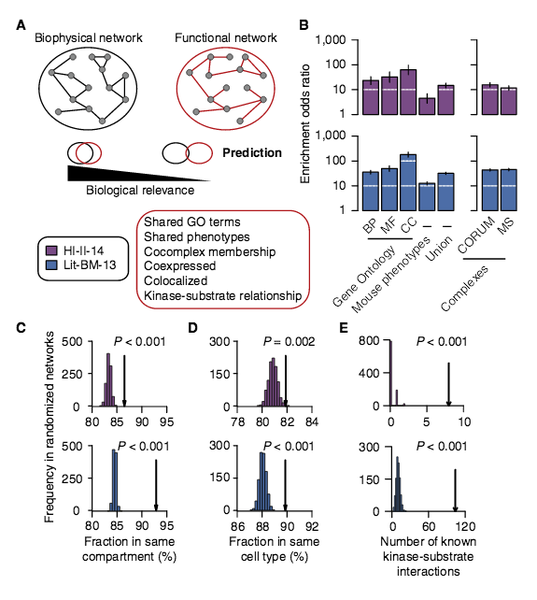
T. Rolland, M. Tasan, , B. Charloteaux, S. J. Pevzner,, Q. Zhong, N. Sahni, S. Yi,, I. Lemmens, C. Fontanillo,, R. Mosca, A. Kamburov, , S. D. Ghiassian, X. Yang,, L. Ghamsari, D. Balcha,, B. E. Begg, P. Braun, M. Brehm, M. P. Froly, A.-R. Carvunis, D, Convery-Zupan, R. Carominas,, J. Coulombe-Huntington, , E. Dann, M. Dreze, A. Dricot,, C. Fan, E. Franzosa, F. Gebrea, B. J. Gutierrez, M. F. Hardy,, M. Jin, S. Kang, R. Kiros, G. , Lin, K. Luck, A. MacWilliams,, J. Menche, R R. Murray, A., Palagi, M. M. Poulin, X. , Rambout, J. Rasla, P. Reichert, V. Romero, E. Ruyssinck, J. M., Sahalie, plus 20 more co-autho
A proteome-scale map of the human interactome network
Cell 159:5, 1212-1226 (2014)
Read the abstract
Just as reference genome sequences revolutionized human genetics, reference maps of interactome networks will be critical to fully understand genotype-phenotype relationships. Here, we describe a systematic map of ∼14,000 high-quality human binary protein-protein interactions. At equal quality, this map is ∼30% larger than what is available from small-scale studies published in the literature in the last few decades. While currently available information is highly biased and only covers a relatively small portion of the proteome, our systematic map appears strikingly more homogeneous, revealing a “broader” human interactome network than currently appreciated. The map also uncovers significant interconnectivity between known and candidate cancer gene products, providing unbiased evidence for an expanded functional cancer landscape, while demonstrating how high-quality interactome models will help “connect the dots” of the genomic revolution.
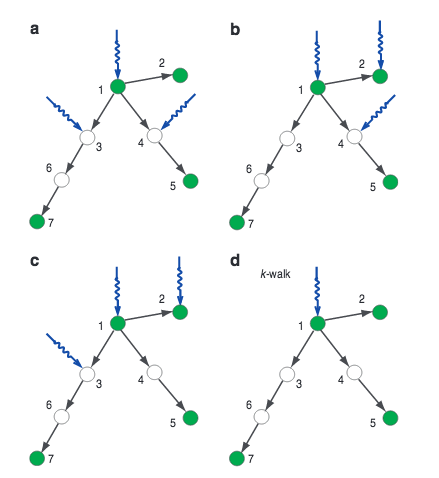
Jianxi Gao, Y.-Y.Liu, R. M. D'Souza, A.-L. Barabási
Target control of complex networks
Nature Communications 5:5415, 1-7 (2014)
Read the abstract
Controlling large natural and technological networks is an outstanding challenge. It is typically neither feasible nor necessary to control the entire network, prompting us to explore target control: the efficient control of a preselected subset of nodes. We show that the structural controllability approach used for full control overestimates the minimum number of driver nodes needed for target control. Here we develop an alternate ‘k-walk’ theory for directed tree networks, and we rigorously prove that one node can control a set of target nodes if the path length to each target node is unique. For more general cases, we develop a greedy algorithm to approximate the minimum set of driver nodes sufficient for target control. We find that degree heterogeneous networks are target controllable with higher efficiency than homogeneous networks and that the structure of many real-world networks are suitable for efficient target control.
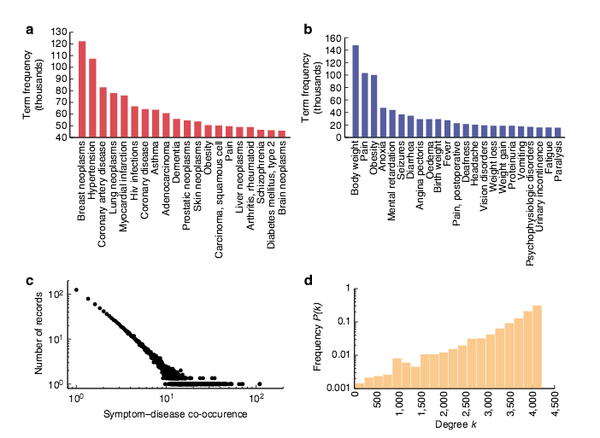
X. Z. Zhou, J. Menche, A.-L. Barabási, A. Sharma
Human symptoms–disease network
Nature Communications 5:4212, 1-10 (2014)
Read the abstract
In the post-genomic era, the elucidation of the relationship between the molecular origins of diseases and their resulting phenotypes is a crucial task for medical research. Here, we use a large-scale biomedical literature database to construct a symptom-based human disease network and investigate the connection between clinical manifestations of diseases and their underlying molecular interactions. We find that the symptom-based similarity of two diseases correlates strongly with the number of shared genetic associations and the extent to which their associated proteins interact. Moreover, the diversity of the clinical manifestations of a disease can be related to the connectivity patterns of the underlying protein interaction network. The comprehensive, high-quality map of disease–symptom relations can further be used as a resource helping to address important questions in the field of systems medicine, for example, the identification of unexpected associations between diseases, disease etiology research or drug design.
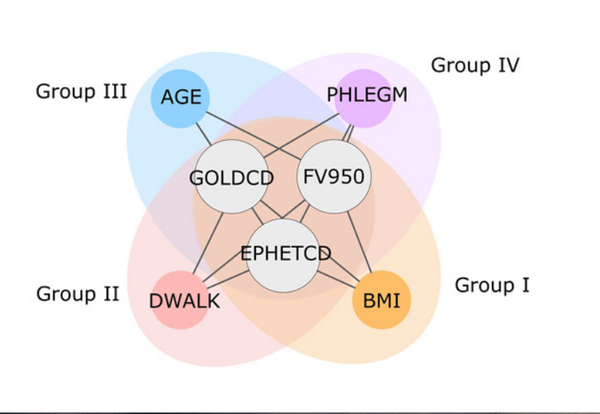
J. Mench, A. Sharma, M. H. Cho, R. J. Mayer, S. I. Rennard, B. Celli, B. E. Miller, N. Locantore, R. Tal-Singer, S. Ghosh, C. Larminie, G. Bradley, J. H. Riley, A. Agusti, E. K. Silverman, A.-L. Barabási
A diVIsive Shuffling Approach (VIStA) for gene expression analysis to identify subtypes in Chronic Obstructive Pulmonary Disease
BMC Systems Biology 8, 1-13 (2014)
Read the abstract
Background: An important step toward understanding the biological mechanisms underlying a complex disease is a refined understanding of its clinical heterogeneity. Relating clinical and molecular differences may allow us to define more specific subtypes of patients that respond differently to therapeutic interventions. Results: We developed a novel unbiased method called diVIsive Shuffling Approach (VIStA) that identifies subgroups of patients by maximizing the difference in their gene expression patterns. We tested our algorithm on 140 subjects with Chronic Obstructive Pulmonary Disease (COPD) and found four distinct, biologically and clinically meaningful combinations of clinical characteristics that are associated with large gene expression differences. The dominant characteristic in these combinations was the severity of airflow limitation. Other frequently identified measures included emphysema, fibrinogen levels, phlegm, BMI and age. A pathway analysis of the differentially expressed genes in the identified subtypes suggests that VIStA is capable of capturing specific molecular signatures within in each group. Conclusions: The introduced methodology allowed us to identify combinations of clinical characteristics that correspond to clear gene expression differences. The resulting subtypes for COPD contribute to a better understanding of its heterogeneity.
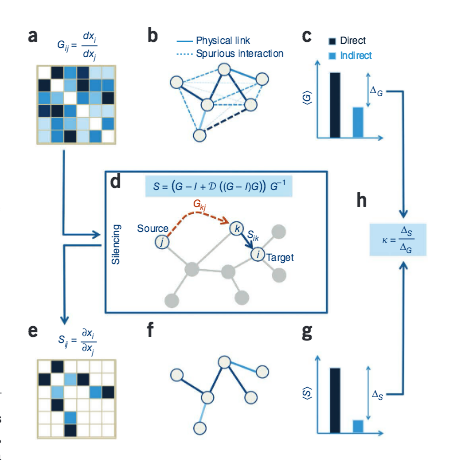
B. Barzel, A.-L. Barabási
Network link prediction by global silencing of indirect correlations
Nature Biotechnology 31: Num 8, 1-8 (2013)
Read the abstract
Predictions of physical and functional links between cellular components are often based on correlations between experimental measurements, such as gene expression. However, correlations are affected by both direct and indirect paths, confounding our ability to identify true pairwise interactions. Here we exploit the fundamental properties of dynamical correlations in networks to develop a method to silence indirect effects. The method receives as input the observed correlations between node pairs and uses a matrix transformation to turn the correlation matrix into a highly discriminative silenced matrix, which enhances only the terms associated with direct causal links. Against empirical data for Escherichia coli regulatory interactions, the method enhanced the discriminative power of the correlations by twofold, yielding >50% predictive improvement over traditional correlation measures and 6% over mutual information. Overall this silencing method will help translate the abundant correlation data into insights about a system's interactions, with applications ranging from link prediction to inferring the dynamical mechanisms governing biological networks.
Y.-Y. Liu, J.-J. Slotine, A.-L. Barabási
Observability of complex systems
Proceedings of the National Academy of Sciences 110, 1-6 (2013)
Read the abstract
A quantitative description of a complex system is inherently limited by our ability to estimate the system’s internal state from experimentally accessible outputs. Although the simultaneous measurement of all internal variables, like all metabolite concentrations in a cell, offers a complete description of a system’s state, in practice experimental access is limited to only a subset of variables, or sensors. A system is called observable if we can reconstruct the system’s complete internal state from its outputs. Here, we adopt a graphical approach derived from the dynamical laws that govern a system to determine the sensors that are necessary to reconstruct the full internal state of a complex system. We apply this approach to biochemical reaction systems, finding that the identified sensors are not only necessary but also sufficient for observability. The developed approach can also identify the optimal sensors for target or partial observability, helping us reconstruct selected state variables from appropriately chosen outputs, a prerequisite for optimal biomarker design. Given the fundamental role observability plays in complex systems, these results offer avenues to systematically explore the dynamics of a wide range of natural, technological and socioeconomic systems.
B. Barzel, A. Sharma, A.-L. Barabási
Graph theory properties of cellular networks (Chapter 9)
Handbook of Systems Biology – Concepts and Insights (Academic Press, Elsevier) , 177-193 (2013)
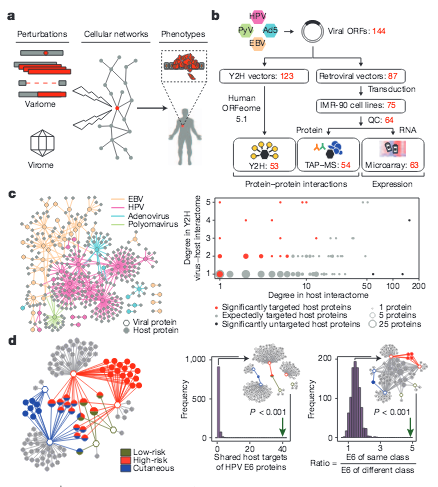
O. Rozenblatt-Rosen, R. C. Deo, M. Padi, G. Adelmant, T. Rolland, M. Grace, A. Dricot, M. Askenazi, M. Tavares, S. J. Pevzner, F. Abderazzaq, D. Byrdsong, A.-R. Carvunis, A. A. Chen, J. Cheng, M. Correll, M. Durate, C. Fan, M. C. Feltkamp, S. B. Ficarro, R. Franchi, B. K. Garg, N. Gulbahce, T. Hao, A. M. Holthaus, R. James, A. Korkhin, L. Litovchick, J. C. Mar, T. R. Pak, S. Rabello, R. Rubio, Y. Shen, S. Singh, J. M. Spangle, M. Tasan, S. Wanamakter, J. T. Webber, J. Roecklein-Canfield,, E. Johannsen, A.-L. Barabasi,, R. Beroukhim, E. Kieff,, M. E. Cusick, D. E. Hill,, K. Munger, J. A. Marto,, J. Quackenbush, F. P. Roth,, J. A. DeCaprio, M. Vidal
Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins
Nature 487, 491-495 (2012)
Read the abstract
Genotypic differences greatly influence susceptibility and resistance to disease. Understanding genotype–phenotype relationships requires that phenotypes be viewed as manifestations of network properties, rather than simply as the result of individual genomic variations. Genome sequencing efforts have identified numerous germline mutations, and large numbers of somatic genomic alterations, associated with a predisposition to cancer. However, it remains difficult to distinguish background, or ‘passenger’, cancer mutations from causal, or ‘driver’, mutations in these data sets. Human viruses intrinsically depend on their host cell during the course of infection and can elicit pathological phenotypes similar to those arising from mutations. Here we test the hypothesis that genomic variations and tumour viruses may cause cancer through related mechanisms, by systematically examining host interactome and transcriptome network perturbations caused by DNA tumour virus proteins. The resulting integrated viral perturbation data reflects rewiring of the host cell networks, and highlights pathways, such as Notch signalling and apoptosis, that go awry in cancer. We show that systematic analyses of host targets of viral proteins can identify cancer genes with a success rate on a par with their identification through functional genomics and large-scale cataloguing of tumour mutations. Together, these complementary approaches increase the specificity of cancer gene identification. Combining systems-level studies of pathogen-encoded gene products with genomic approaches will facilitate the prioritization of cancer causing driver genes to advance the understanding of the genetic basis of human cancer.
J. Loscalzo, A.-L. Barabási
Systems biology and the future of medicine
WIREs Systems Biology and Medicine 3, 619-627 (2011)
Read the abstract
Contemporary views of human disease are based on simple correlation between clinical syndromes and pathological analysis dating from the late 19th century. Although this approach to disease diagnosis, prognosis, and treatment has served the medical establishment and society well for many years, it has serious shortcomings for the modern era of the genomic medicine that stem from its reliance on reductionist principles of experimentation and analysis. Quantitative, holistic systems biology applied to human disease offers a unique approach for diagnosing established disease, defining disease predilection, and developing individualized (personalized) treatment strategies that can take full advantage of modern molecular pathobiology and the comprehensive data sets that are rapidly becoming available for populations and individuals. In this way, systems pathobiology offers the promise of redefining our approach to disease and the field of medicine.
Y.-Y. Liu, J.-J. Slotine, A.-L. Barabási
Few inputs can reprogram biological networks (reply by Liu et al.)
Nature 473, 167-173 (2011)
Read the abstract
Reply to Franz-Josef Muller and Andreas Schuppert (Nature 478, Pg. E4, Oct. 2011)
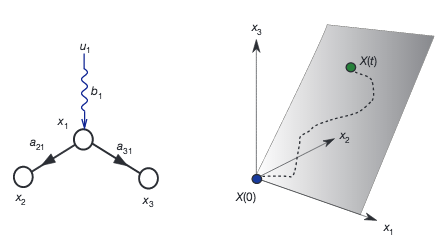
Y.-Y. Liu, J.-J. Slotine, A.-L. Barabási
Controllability of complex networks
Nature 473, 167-173 (2011)
Read the abstract
The ultimate proof of our understanding of natural or technological systems is reflected in our ability to control them. Although control theory offers mathematical tools for steering engineered and natural systems towards a desired state, a framework to control complex self-organized systems is lacking. Here we develop analytical tools to study the controllability of an arbitrary complex directed network, identifying the set of driver nodes with time-dependent control that can guide the system’s entire dynamics. We apply these tools to several real networks, finding that the number of driver nodes is determined mainly by the network’s degree distribution. We show that sparse inhomogeneous networks, which emerge in many real complex systems, are the most difficult to control, but that dense and homogeneous networks can be controlled using a few driver nodes. Counterintuitively, we find that in both model and real systems the driver nodes tend to avoid the high-degree nodes.
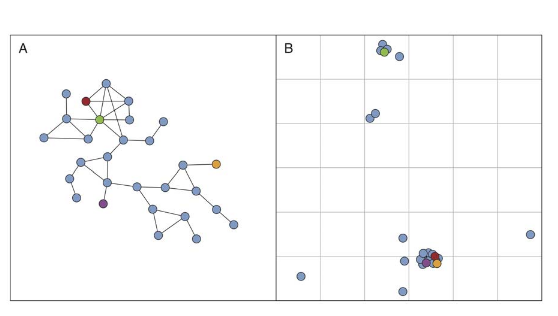
J. P. Onnela, S. Arbesman, M. C. Gonzalez, A.-L. Barabasi, N. A. Christakis
Geographic Constraints on Social Network Groups
PLoS One 6:4, 1-7 (2011)
Read the abstract
Social groups are fundamental building blocks of human societies. While our social interactions have always been constrained by geography, it has been impossible, due to practical difficulties, to evaluate the nature of this restriction on social group structure. We construct a social network of individuals whose most frequent geographical locations are also known. We also classify the individuals into groups according to a community detection algorithm. We study the variation of geographical span for social groups of varying sizes, and explore the relationship between topological positions and geographic positions of their members. We find that small social groups are geographically very tight, but become much more clumped when the group size exceeds about 30 members. Also, we find no correlation between the topological positions and geographic positions of individuals within network communities. These results suggest that spreading processes face distinct structural and spatial constraints.
M. Vidal, M. E. Cusick, A.-L. Barabasi
Interactome Networks and Human Disease
Cell 144, 986-995 (2011)
Read the abstract
Complex biological systems and cellular networks may underlie most genotype to phenotype relationships. Here, we review basic concepts in network biology, discussing different types of interactome networks and the insights that can come from analyzing them. We elaborate on why interactome networks are important to consider in biology, how they can be mapped and integratedwith each other, what global properties are starting to emerge from interactome network models, and how these properties may relate to human disease.
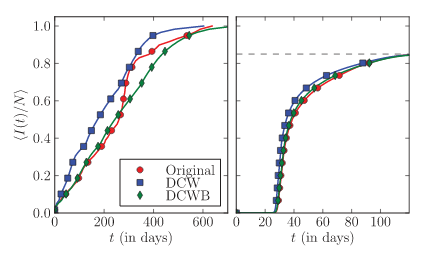
M. Karsai, M. Kivelä, R. K. Pan, K. Kaski, J. Kertész, A.-L. Barabási, J. Saramäki
Small but slow world: How network topology and burstiness slow down spreading
Physical Review E 83, 1-4 (2011)
Read the abstract
While communication networks show the small-world property of short paths, the spreading dynamics in them turns out slow. Here, the time evolution of information propagation is followed through communication networks by using empirical data on contact sequences and the susceptible-infected model. Introducing null models where event sequences are appropriately shuffled, we are able to distinguish between the contributions of different impeding effects. The slowing down of spreading is found to be caused mainly by weight-topology correlations and the bursty activity patterns of individuals.
J. J. Kahle, N. Gulbahce, C. A. Shaw, J. Lim, D. E. Hill, A.-L. Barabás, H. Y. Zoghbi
Comparison of an expanded ataxia interactome with patient medical records reveals a relationship between macular degeneration and ataxia
Human Molecular Genetics 20, 510-527 (2011)
Read the abstract
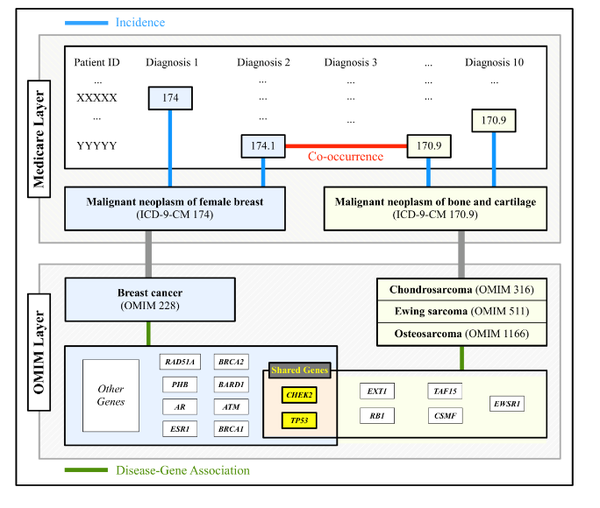
Spinocerebellar ataxias 6 and 7 (SCA6 and SCA7) are neurodegenerative disorders caused by expansion of CAG repeats encoding polyglutamine (polyQ) tracts in CACNA1A, the alpha1A subunit of the P/Q-type calcium channel, and ataxin-7 (ATXN7), a component of a chromatin-remodeling complex, respectively. We hypothesized that finding new protein partners for ATXN7 and CACNA1A would provide insight into the biology of their respective diseases and their relationship to other ataxia-causing proteins. We identified 118 protein interactions for CACNA1A and ATXN7 linking them to other ataxia-causing proteins and the ataxia network. To begin to understand the biological relevance of these protein interactions within the ataxia network, we used OMIM to identify diseases associated with the expanded ataxia network. We then used Medicare patient records to determine if any of these diseases co-occur with hereditary ataxia. We found that patients with ataxia are at 3.03-fold greater risk of these diseases than Medicare patients overall. One of the diseases comorbid with ataxia is macular degeneration (MD). The ataxia network is significantly (P= 7.37 × 10(-5)) enriched for proteins that interact with known MD-causing proteins, forming a MD subnetwork. We found that at least two of the proteins in the MD subnetwork have altered expression in the retina of Ataxin-7(266Q/+) mice suggesting an in vivo functional relationship with ATXN7. Together these data reveal novel protein interactions and suggest potential pathways that can contribute to the pathophysiology of ataxia, MD, and diseases comorbid with ataxia.
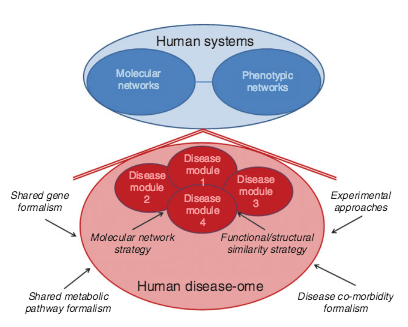
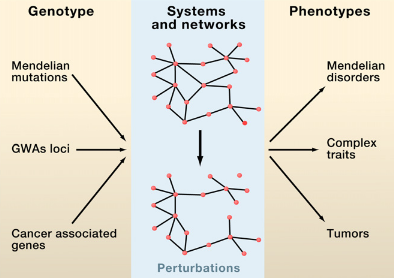
A.-L. Barabási, N. Gulbahce, J. Loscalzo
Network medicine: a network-based approach to human disease
Nature Reviews Genetics 12, 56-68 (2011)
Read the abstract
Given the functional interdependencies between the molecular components in a human cell, a disease is rarely a consequence of an abnormality in a single gene, but reflects the perturbations of the complex intracellular and intercellular network that links tissue and organ systems. The emerging tools of network medicine offer a platform to explore systematically not only the molecular complexity of a particular disease, leading to the identification of disease modules and pathways, but also the molecular relationships among apparently distinct (patho)phenotypes. Advances in this direction are essential for identifying new disease genes, for uncovering the biological significance of disease-associated mutations identified by genome-wide association studies and full-genome sequencing, and for identifying drug targets and biomarkers for complex diseases.
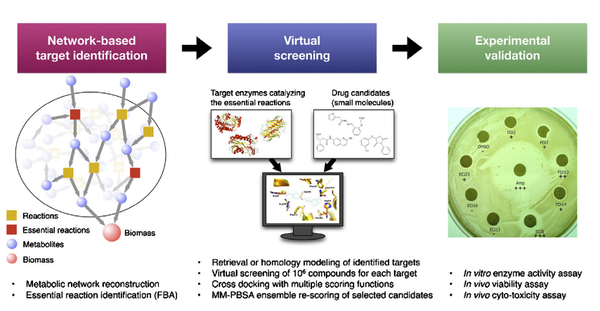
Y. Shen, L. Liu, G. Estiu, B. Isin, Y.-Y. Ahn, D.-S. Lee, A.-L. Barabásii, v. Kapatral, O. Wiest, Z. N. Oltvai
Blueprint for antimicrobial hit discovery targeting metabolic networks
Proceedings of the National Academy of Sciences of the United States of America 10.1073, 1-6 (2010)
Read the abstract
Advances in genome analysis, network biology, and computational chemistry have the potential to revolutionize drug discovery by combining system-level identification of drug targets with the atomistic modeling of small molecules capable of modulating their activity. To demonstrate the effectiveness of such a discovery pipeline, we deduced common antibiotic targets in Escherichia coli and Staphylococcus aureus by identifying shared tissue-specific or uniformly essential metabolic reactions in their metabolic networks. We then predicted through virtual screening dozens of potential inhibitors for several enzymes of these reactions and showed experimentally that a subset of these inhibited both enzyme activities in vitro and bacterial cell viability. This blueprint is applicable for any sequenced organism with high-quality metabolic reconstruction and suggests a general strategy for strain-specific antiinfective therapy.
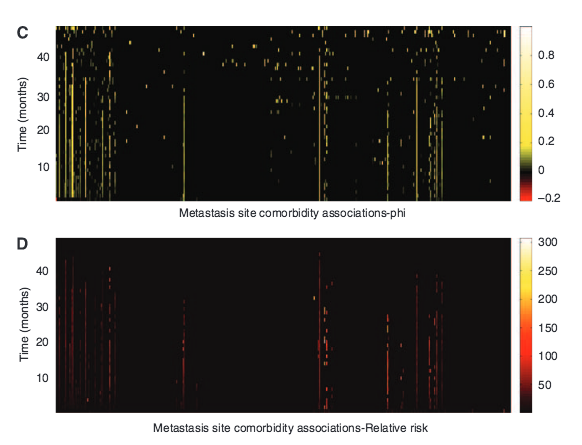
L. L. Chen, N. Blumm, N. A. Christakis, A.-L. Barabási, T. S. Deisboeck
Cancer metastasis networks and the prediction of progression patterns
British Journal of Cancer 101, 749-758 (2009)
Read the abstract
Background: Metastasis patterns in cancer vary both spatially and temporally. Network modeling may allow the incorporation of the temporal dimension in the analysis of these patterns.METHODS: We used Medicare claims of 2 265 167 elderly patients aged X65 years to study the large-scale clinical pattern of metastases. We introduce the concept of a cancer metastasis network, in which nodes represent the primary cancer site and the sites of subsequent metastases, connected by links that measure the strength of co-occurrence.RESULTS: These cancer metastasis networks capture both temporal and subtle relational information, the dynamics of which differ between cancer types. Using these networks as entities on which the metastatic disease of individual patients may evolve, we show that they may be used, for certain cancer types, to make retrograde predictions of a primary cancer type given a sequence ofmetastases, as well as anterograde predictions of future sites of metastasis.
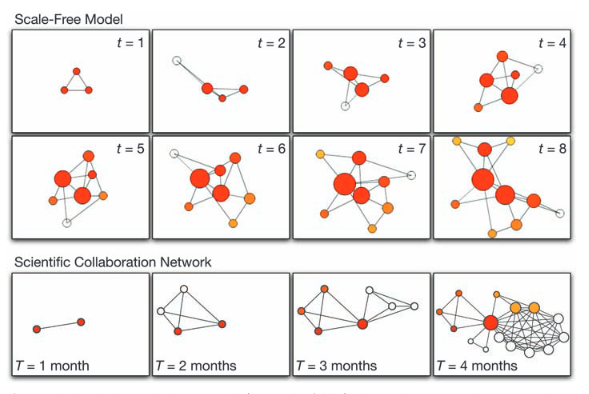
A.-L. Barabási
Scale-Free Networks: A Decade and Beyond
Science 325, 412-413 (2009)
Read the abstract
For decades, we tacitly assumed that the components of such complex systems as the cell, the society, or the Internet are randomly wired together. In the past decade, an avalanche of research has shown that many real networks, independent of their age, function, and scope, converge to similar architectures, a universality that allowed researchers from different disciplines to embrace network theory as a common paradigm. The decade-old discovery of scale-free networks was one of those events that had helped catalyze the emergence of network science, a new research field with its distinct set of challenges and accomplishments.
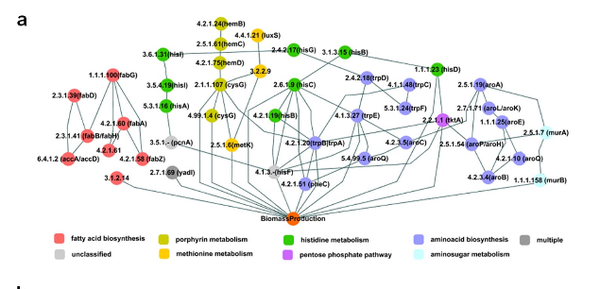
D.-S. Lee, H. Burd, J. Liu, E. Almass, O. Weist, A.-L. Barabási, Z. N. Oltvai, V. Kapatra
Comparative Genome-Scale Metabolic Reconstruction and Flux Balance Analysis of Multiple Staphylococcus aureus Genomes Identify Novel Antimicrobial Drug Targets
Journal of Bacteriology 191:12, 4015–4024 (2009)
Read the abstract
Mortality due to multidrug-resistant Staphylococcus aureus infection is predicted to surpass that of human immunodeficiency virus/AIDS in the United States. Despite the various treatment options for S. aureus infections, it remains a major hospital- and community-acquired opportunistic pathogen. With the emergence of multidrug-resistant S. aureus strains, there is an urgent need for the discovery of new antimicrobial drug targets in the organism. To this end, we reconstructed the metabolic networks of multidrug-resistant S. aureus strains using genome annotation, functional-pathway analysis, and comparative genomic approaches, followed by flux balance analysis-based in silico single and double gene deletion experiments. We identified 70 single enzymes and 54 pairs of enzymes whose corresponding metabolic reactions are predicted to be unconditionally essential for growth. Of these, 44 single enzymes and 10 enzyme pairs proved to be common to all 13 S. aureus strains, including many that had not been previously identified as being essential for growth by gene deletion experiments in S. aureus. We thus conclude that metabolic reconstruction and in silico analyses of multiple strains of the same bacterial species provide a novel approach for potential antibiotic target identification.
C. A. Hidalgo, N. Blumm, A.-L. Barabási, N. A. Christakis
A dynamic network approach for the study of human phenotypes
PLoS Computational Biology 5:4, 1-11 (2009)
Read the abstract
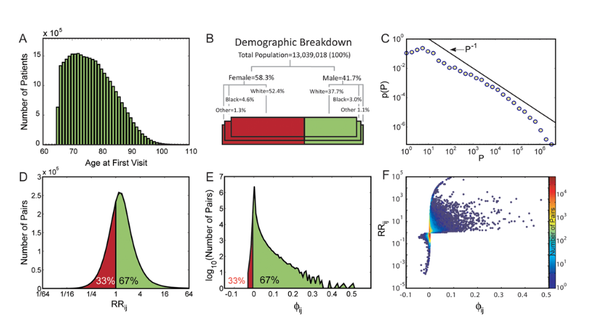
The use of networks to integrate different genetic, proteomic, and metabolic datasets has been proposed as a viable path toward elucidating the origins of specific diseases. Here we introduce a new phenotypic database summarizing correlations obtained from the disease history of more than 30 million patients in a Phenotypic Disease Network (PDN). We present evidence that the structure of the PDN is relevant to the understanding of illness progression by showing that (1) patients develop diseases close in the network to those they already have; (2) the progression of disease along the links of the network is different for patients of different genders and ethnicities; (3) patients diagnosed with diseases which are more highly connected in the PDN tend to die sooner than those affected by less connected diseases; and (4) diseases that tend to be preceded by others in the PDN tend to be more connected than diseases that precede other illnesses, and are associated with higher degrees of mortality. Our findings show that disease progression can be represented and studied using network methods, offering the potential to enhance our understanding of the origin and evolution of human diseases. The dataset introduced here, released concurrently with this publication, represents the largest relational phenotypic resource publicly available to the research community.
J. Park, D. S. Lee, N. A. Christakis, A.-L. Barabási
The impact of cellular networks on disease comorbidity
Molecular Systems Biology 5:262, 1-7 (2009)
Read the abstract
The impact of disease-causing defects is often not limited to the products of a mutated gene but, thanks to interactions between the molecular components, may also affect other cellular functions, resulting in potential comorbidity effects. By combining information on cellular interactions, disease--gene associations, and population-level disease patterns extracted from Medicare data, we find statistically significant correlations between the underlying structure of cellular networks and disease comorbidity patterns in the human population. Our results indicate that such a combination of population-level data and cellular network information could help build novel hypotheses about disease mechanisms.
A. Vazquez, M. A. de Menezes, A.-L. Barabási, Z. N. Oltvai
Impact of Limited Solvent Capacity on Metabolic Rate, Enzyme Activities, and Metabolite Concentrations of S.cerevisiae Glycolysis
PLoS Computational Biology 4:10, 1-6 (2008)
Read the abstract
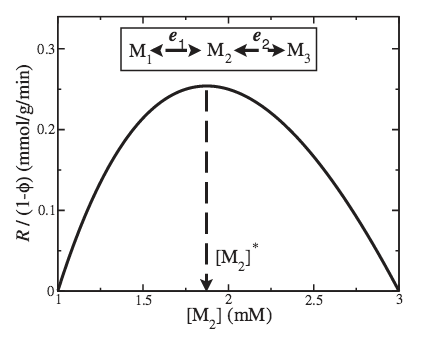
The cell’s cytoplasm is crowded by its various molecular components, resulting in a limited solvent capacity for the allocation of new proteins, thus constraining various cellular processes such as metabolism. Here we study the impact of the limited solvent capacity constraint on the metabolic rate, enzyme activities, and metabolite concentrations using a computational model of Saccharomyces cerevisiae glycolysis as a case study. We show that given the limited solvent capacity constraint, the optimal enzyme activities and the metabolite concentrations necessary to achieve a maximum rate of glycolysis are in agreement with their experimentally measured values. Furthermore, the predicted maximum glycolytic rate determined by the solvent capacity constraint is close to that measured in vivo. These results indicate that the limited solvent capacity is a relevant constraint acting on S. cerevisiae at physiological growth conditions, and that a full kinetic model together with the limited solvent capacity constraint can be used to predict both metabolite concentrations and enzyme activities in vivo.
H. Yu, P. Braun, M. A. Yildirim, I. Lemmens, K. Venkatesan, J. Sahalie, T. Hirozane-Kishikawa, F. Gebreab, N. Li, N. Simonis, T. Hao, J.-F. Raul, A. Dricot, A. Vazquez, R. R. Murray, C. Simon, L. Tardivo, S. Tam, N. Svrzikapa, C. Fan, A.-S. de Semt, A. Motyl, M. E. Hudson, J. Park, X. Xin, M. E. Cusick, T. Moore, C. Boone, M. Snyder, F. P. Roth, A.-L. Barabási, J. Tavernier, D. E. Hill, M. Vidal
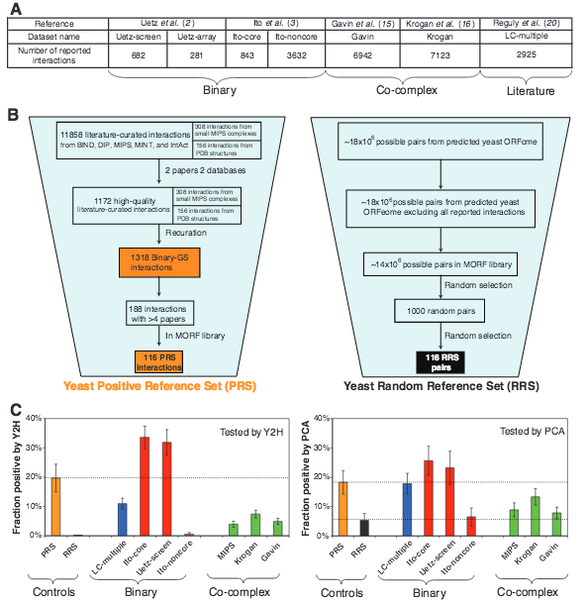
High-Quality Binary Protein Interaction Map of the Yeast Interactome Network
Science 322, 104-110 (2008)
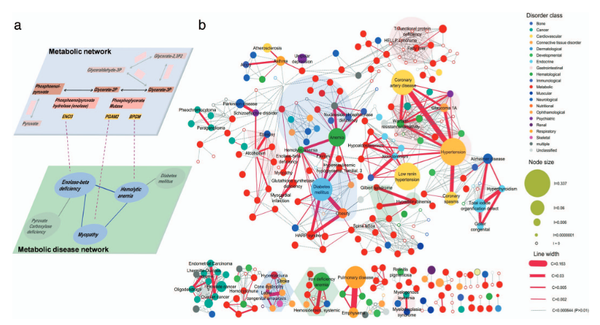
D.-S. Lee, J. Park, K. A. Kay, N. A. Christakis, Z. N. Oltvai, A.-L. Barabási
The implications of human metabolic network topology for disease comorbidity
Proceedings of the National Academy of Sciences 105, 9880-9885 (2008)
Read the abstract
Most diseases are the consequence of the breakdown of cellular processes, but the relationships among genetic/epigenetic defects, the molecular interaction networks underlying them, and the disease phenotypes remain poorly understood. To gain insights into such relationships, here we constructed a bipartite human disease association network in which nodes are diseases and two diseases are linked if mutated enzymes associated with them catalyze adjacent metabolic reactions. We find that connected disease pairs display higher correlated reaction flux rate, corresponding enzyme-encoding gene coexpression, and higher comorbidity than those that have no metabolic link between them. Furthermore, the more connected a disease is to other diseases, the higher is its prevalence and associated mortality rate. The network topology-based approach also helps to
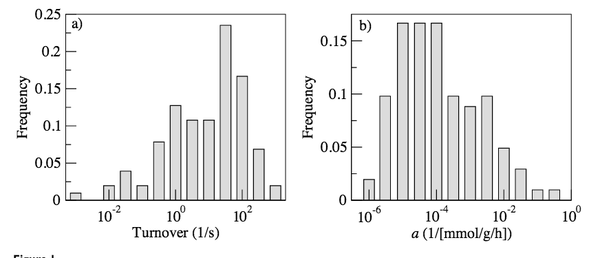
A. Vazquez, Q. K. Beg, M. A. de Menezes, J. Ernst, Z. Bar-Joseph, A.-L. Barabási, L. G. Boros, Z. N. Oltvai
Impact of the solvent capacity constraint on E. coli metabolism
BMC Systems Biology 2:7, 1-10 (2008)
Read the abstract
Obtaining quantitative predictions for cellular metabolic activities requires the identification and modeling of the physicochemical constraints that are relevant at physiological growth conditions. Molecular crowding in a cell's cytoplasm is one such potential constraint, as it limits the solvent capacity available to metabolic enzymes.
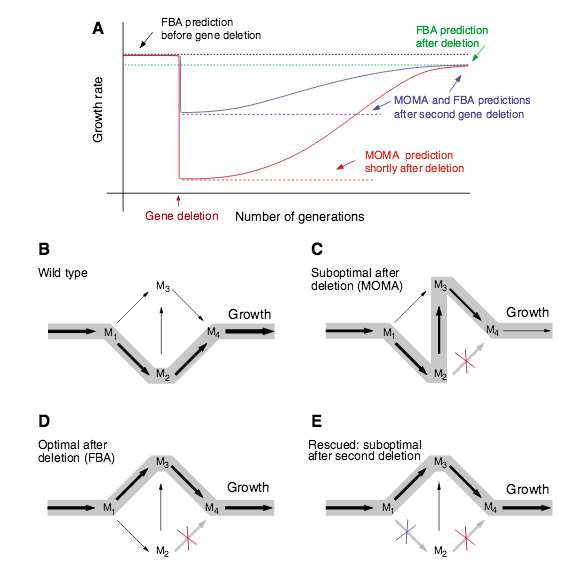
A. Motter, N. Gulbahce, E. Almaas, A.-L. Barabási
Predicting synthetic rescues in metabolic networks
Molecular Systems Biology 4:168, 1-10 (2008)
Read the abstract
An important goal of medical research is to develop methods to recover the loss of cellular function due to mutations and other defects. Many approaches based on gene therapy aim to repair the defective gene or to insert genes with compensatory function. Here, we propose an alternative, network-based strategy that aims to restore biological function by forcing the cell to either bypass the functions affected by the defective gene, or to compensate for the lost function. Focusing on the metabolism of single-cell organisms, we computationally study mutants that lack an essential enzyme, and thus are unable to grow or have a significantly reduced growth rate. We show that several of these mutants can be turned into viable organisms through additional gene deletions that restore their growth rate. In a rather counterintuitive fashion, this is achieved via additional damage to the metabolic network. Using flux balance-based approaches, we identify a number of synthetically viable gene pairs, in which the removal of one enzyme-encoding gene results in a non-viable phenotype, while the deletion of a second enzyme-encoding gene rescues the organism. The systematic network-based identification of compensatory rescue effects may open new avenues for genetic interventions.
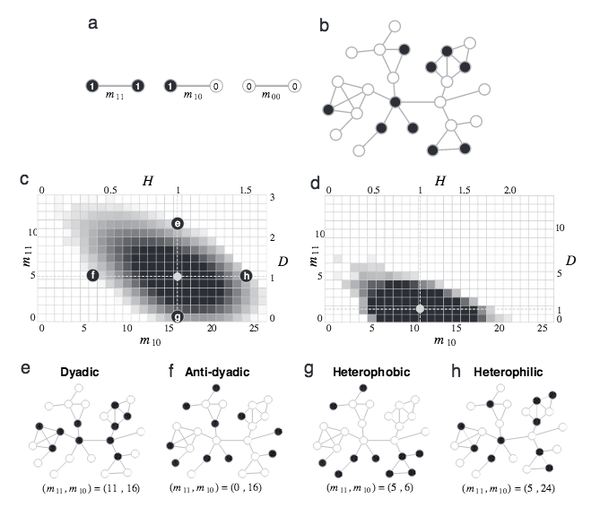
J. Park, A-L. Barabási
Distribution of node characteristics in complex networks
Proceedings of the National Academy of Sciences 104, 17916-17920 (2007)
Read the abstract
Our enhanced ability to map the structure of various complex networks is increasingly accompanied by the possibility of independently identifying the functional characteristics of each node. Although this led to the observation that nodes with similar characteristics have a tendency to link to each other, in general we lack the tools to quantify the interplay between node properties and the structure of the underlying network. Here we show that when nodes in a network belong to two distinct classes, two independent parameters are needed to capture the detailed interplay between the network structure and node properties. We find that the network structure significantly limits the values of these parameters, requiring a phase diagram to uniquely characterize the configurations available to the system. The phase diagram shows a remarkable independence from the network size, a finding that, together with a proposed heuristic algorithm, allows us to determine its shape even for large networks. To test the usefulness of the developed methods, we apply them to biological and socioeconomic systems, finding that protein functions and mobile phone usage occupy distinct regions of the phase diagram, indicating that the proposed parameters have a strong discriminating power.
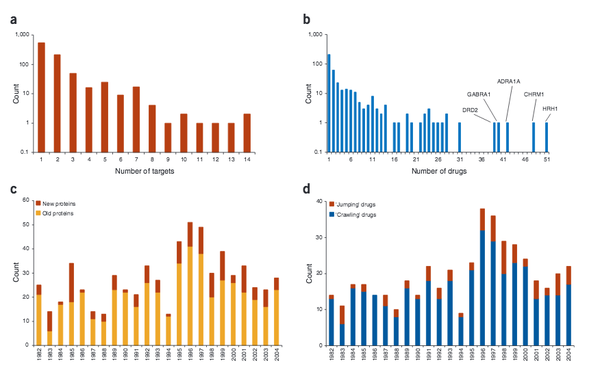
M. A. Yildirim, K.-L. Goh, M.E. Cusick, A.-L. Barabási, M. Vidal
Drug-target network
Nature Biotechnology 25:10, 1119-1126 (2007)
Read the abstract
The global set of relationships between protein targets of all drugs and all disease-gene products in the human protein–protein interaction or ‘interactome’ network remains uncharacterized. We built a bipartite graph composed of US Food and Drug Administration–approved drugs and proteins linked by drug–target binary associations. The resultingnetwork connects most drugs into a highly interlinked giant component, with strong local clustering of drugs of similar types according to Anatomical Therapeutic Chemical classification. Topological analyses of this network quantitatively showed an overabundance of ‘follow-on’ drugs, that is, drugs that target already targeted proteins. By including drugs currently under investigation, we identified a trend toward more functionally diverse targets improving polypharmacology. To analyze the relationships between drug targets and disease-gene products, we measured the shortest distance between both sets of proteins in current models of the human interactome network. Significant differences in distance were found between etiological and palliative drugs. A recent trend toward more rational drug design was observed.
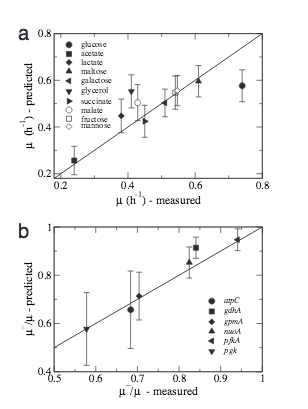
Q. K. Beg, A. Vazquez, J. Ernst, M. A. de Menezes, Z. Bar-Joseph, A.-L. Barabási, Z. N. Oltvai
Intracellular crowding defines the mode and sequence of substrate uptake by Escherichia coli and constrains its metabolic activity
Proceedings of the National Academy of Sciences 104, 31 (2007)
Read the abstract
The influence of the high intracellular concentration of macromolecules on cell physiology is increasingly appreciated, but its impact on system-level cellular functions remains poorly quantified. To assess its potential effect, here we develop a flux balance model of Escherichia coli cell metabolism that takes into account a systemslevel constraint for the concentration of enzymes catalyzing the various metabolic reactions in the crowded cytoplasm. We demonstrate that the model’s predictions for the relative maximum growth rate of wild-type and mutant E. coli cells in single substratelimited media, and the sequence and mode of substrate uptake and utilization from a complex medium are in good agreement with subsequent experimental observations. These results suggest that molecular crowding represents a bound on the achievable functional states of a metabolic network, and they indicate that models incorporating this constraint can systematically identify alterations in cellular metabolism activated in response to environmental change.
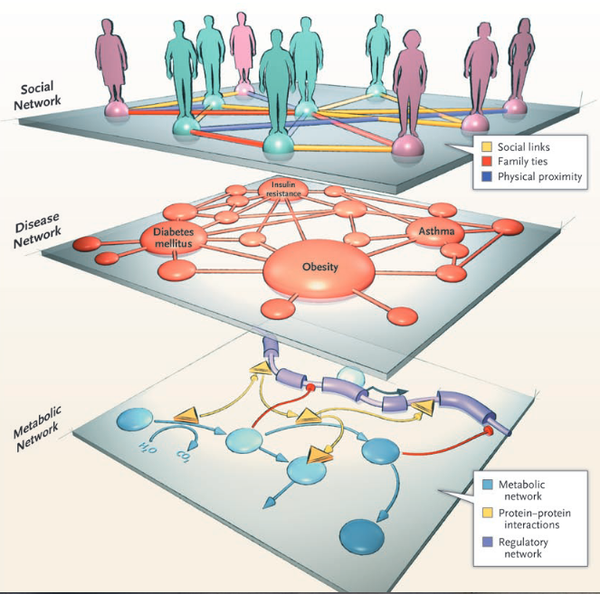
A.L. Barabási
Network Medicine — From Obesity to the “Diseasome”
New England Journal of Medicine 357, 404-407 (2007)
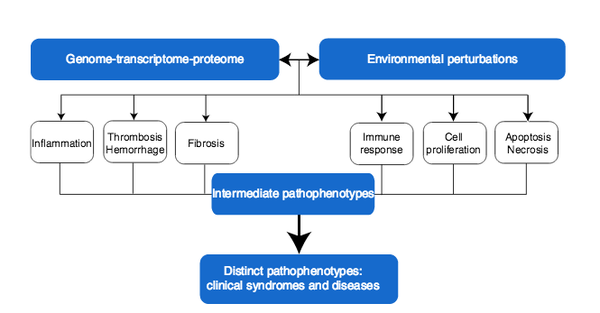
J. Loscalzo, I. Kohane, A.-L. Barabási
Human disease classification in the postgenomic era: A complex systems approach to human pathobiology
Molecular Systems Biology 3:124, 1-11 (2007)
Read the abstract
Contemporary classification of human disease derives from observational correlation between pathological analysis and clinical syndromes. Characterizing disease in this way established a nosology that has served clinicians well to the current time, and depends on observational skills and simple laboratory tools to define the syndromic phenotype. Yet, this time-honored diagnostic strategy has significant shortcomings that reflect both a lack of sensitivity in identifying preclinical disease, and a lack of specificity in defining disease unequivocally. In this paper, we focus on the latter limitation, viewing it as a reflection both of the different clinical presentations of many diseases (variable phenotypic expression), and of the excessive reliance on Cartesian reductionism in establishing diagnoses. The purpose of this perspective is to provide a logical basis for a new approach to classifying human disease that uses.

V. Vermeirssen, M. Inmaculada Barrasa, C. Hidalgo, J.-A. B. Babon, R. Sequerra, L. Doucette-Stamm, A.-L. Barabási, A. J.M. Walhout
Transcription factor modularity in a Gene-Centered C. elegans Protein-DNA interaction network
Genome Research 17, 061-1071 (2007)
Read the abstract
Transcription regulatory networks play a pivotal role in the development, function, and pathology of metazoan organisms. Such networks are comprised of protein–DNA interactions between transcription factors (TFs) and their target genes. An important question pertains to how the architecture of such networks relates to network functionality. Here, we show that a Caenorhabditis elegans core neuronal protein–DNA interaction network is organized into two TF modules. These modules contain TFs that bind to a relatively small number of target genes and are more systems specific than the TF hubs that connect the modules. Each module relates to different functional aspects of the network. One module contains TFs involved in reproduction and target genes that are expressed in neurons as well as in other tissues. The second module is enriched for paired homeodomain TFs and connects to target genes.
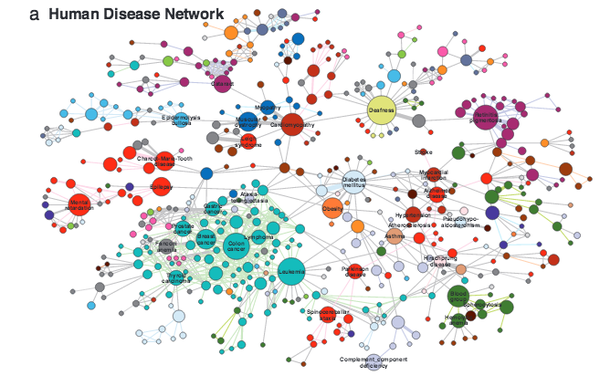
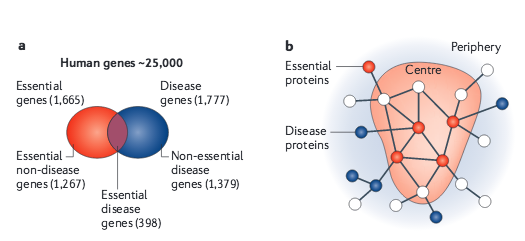
K.-I. Goh, M. E. Cusick, D. Valle, B. Childs, M. Vidal, A.-L. Barabási
The human disease network
Proceedings of the National Academy of Sciences 104:21, 8685 (2007)
Read the abstract
A network of disorders and disease genes linked by known disorder–gene associations offers a platform to explore in a single graphtheoretic framework all known phenotype and disease gene associations, indicating the common genetic origin of many diseases. Genes associated with similar disorders show both higher likelihood of physical interactions between their products and higher expression profiling similarity for their transcripts, supporting the existence of distinct disease-specific functional modules. We find that essential human genes are likely to encode hub proteins and are expressed widely in most tissues. This suggests that disease genes also would play a central role in the human interactome. In contrast, we find that the vast majority of disease genes are nonessential and show no tendency to encode hub proteins, and their expression pattern indicates that they are localized in the functional periphery of the network. A selection-based model explains the observed difference between essential and disease genes and also suggests that diseases caused by somatic mutations should not be peripheral, a prediction we confirm for cancer genes.
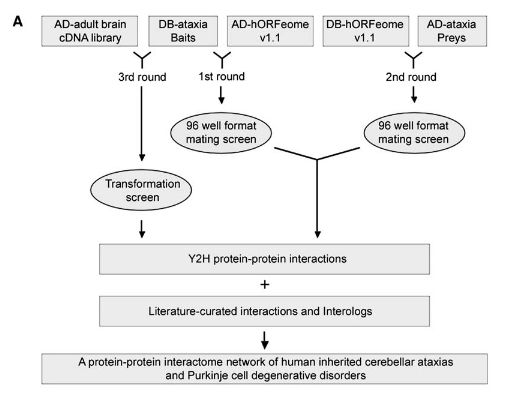
J. Lim, T. Hao, C. Shaw, A.J. Patel, G. Szabo, J.F. Rual, C.J. Fisk, N. Li, A. Smolyar, D.E. Hill, A.-L. Barabási, M. Vidal, H.Y. Zoghbi
A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration
Cell 125, 801-814 (2006)
Read the abstract
Many human inherited neurodegenerative disorders are characterized by loss of balance due to cerebellar Purkinje cell (PC) degeneration. Although the disease-causing mutations have been identified for a number of these disorders, the normal functions of the proteins involved remain, in many cases, unknown. To gain insight into the function of proteins involved in PC degeneration, we developed an interaction network for 54 proteins involved in 23 inherited ataxias and expanded the network by incorporating literature-curated and evolutionarily conserved interactions. We identified 770 mostly novel protein–protein interactions using a stringent yeast two-hybrid screen; of 75 pairs tested, 83% of the interactions were verified in mammalian cells. Many ataxia-causing proteins share interacting partners, a subset of which have been found to modify neurodegeneration in animal models. This interactome thus provides a tool for understanding pathogenic mechanisms common for this class of neurodegenerative disorders and for identifying candidate genes for inherited ataxias.
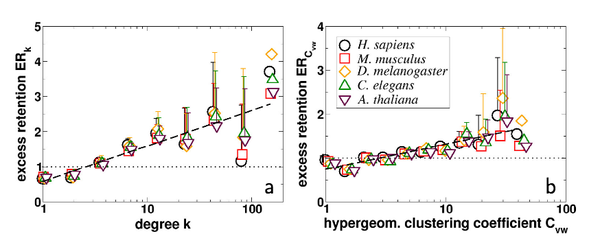
S. Wuchty, A.-L. Barabási, M.T. Ferdig
Stable evolutionary signal in a Yeast protein interaction network
BMC Evolutionary Biology 60, 8 (2006)
Read the abstract
Background: The recently emerged protein interaction network paradigm can provide novel and important insights into the innerworkings of a cell. Yet, the heavy burden of both false positive and false negative protein-protein interaction data casts doubt on the broader usefulness of these interaction sets. Approaches focusing on one-protein-at-a-time have been powerfully employed to demonstrate the high degree of conservation of proteins participating in numerous interactions; here, we expand his 'node' focused paradigm to investigate the relative persistence of 'link' based evolutionary signals in a protein interaction network of S. cerevisiae and point out the value of this relatively untapped source of information. Results: The trend for highly connected proteins to be preferably conserved in evolution is stable, even in the context of tremendous noise in the underlying protein interactions as well as in the assignment of orthology among five higher eukaryotes. We find that local clustering around interactions correlates with preferred evolutionary conservation of the participating proteins; furthermore the correlation between high local clustering and evolutionary conservation is accompanied by a stable elevated degree of coexpression of the interacting proteins. We use this conserved interaction data, combined with P. falciparum /Yeast orthologs, as proof-of-principle that high-order network topology can be used comparatively to deduce local network structure in nonmodel organisms. Conclusion: High local clustering is a criterion for the reliability of an interaction and coincides with preferred evolutionary conservation and significant coexpression. These strong and stable correlations indicate that evolutionary units go beyond a single protein to include the interactions among them. In particular, the stability of these signals in the face of extreme noise suggests that empirical protein interaction data can be integrated with orthologous clustering around these protein interactions to reliably infer local network structures in non-model organisms.

E. Almaas, Z.N. Oltvai, A.-L. Barabási
The activity reaction core and plasticity of metabolic networks
PLOS Computational Biology 1, 557-563 (2005)
Read the abstract
Understanding the system-level adaptive changes taking place in an organism in response to variations in the environment is a key issue of contemporary biology. Current modeling approaches, such as constraint-based fluxbalance analysis, have proved highly successful in analyzing the capabilities of cellular metabolism, including its capacity to predict deletion phenotypes, the ability to calculate the relative flux values of metabolic reactions, and the capability to identify properties of optimal growth states. Here, we use flux-balance analysis to thoroughly assess the activity of Escherichia coli, Helicobacter pylori, and Saccharomyces cerevisiae metabolism in 30,000 diverse simulated environments. We identify a set of metabolic reactions forming a connected metabolic core that carry non-zero fluxes under all growth conditions, and whose flux variations are highly correlated. Furthermore, we find that the enzymes catalyzing the core reactions display a considerably higher fraction of phenotypic essentiality and evolutionary conservation than those catalyzing noncore reactions. Cellular metabolism is characterized by a large number of species-specific conditionally active reactions organized around an evolutionary conserved, but always active, metabolic core. Finally, we find that most current antibiotics interfering with bacterial metabolism target the core enzymes, indicating that our findings may have important implications for antimicrobial drug-target discovery.
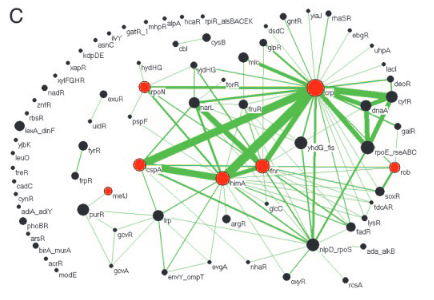
G. Balazsi, A.-L. Barabási, Z.N. Oltvai
Topological units of environmental signal processing in the transcriptional regulatory network of Escherichia coli
Proceedings of the National Academy of Sciences 102, 7841-7846 (2005)
Read the abstract
Recent evidence indicates that potential interactions within metabolic, protein–protein interaction, and transcriptional regulatory networks are used differentially according to the environmental conditions in which a cell exists. However, the topological units underlying such differential utilization are not understood. Here we use the transcriptional regulatory network of Escherichia coli to identify such units, called origons, representing regulatory subnetworks that originate at a distinct class of sensor transcription factors. Using microarray data, we find that specific environmental signals affect mRNA expression levels significantly only within the origons responsible for their detection and processing. We also show that small regulatory interaction patterns, called subgraphs and motifs, occupy distinct positions in and between origons, offering insights into their dynamical role in information processing. The identified features are likely to represent a general framework for environmental signal processing in prokaryotes.
A. Vazquez, R. Dobrin, D. Sergi, J.-P. Eckmann, Z. N. Oltvai, A.-L. Barabási
The topological relationship between the large-scale attributes and local interactions patterns of complex networks
Proceedings of the National Academy of Sciences 101, 17940-17945 (2004)
Read the abstract
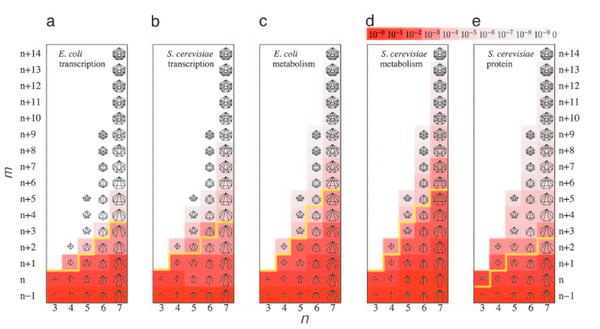
Recent evidence indicates that the abundance of recurring elementary interaction patterns in complex networks, often called subgraphs or motifs, carry significant information about their function and overall organization. Yet, the underlying reasons for the variable quantity of different subgraph types, their propensity to form clusters, and their relationship with the networks’ global organization remain poorly understood. Here we show that a network’s large-scale topological organization and its local subgraph structure mutually define and predict each other, as confirmed by direct measurements in five well studied cellular networks. We also demonstrate the inherent existence of two distinct classes of subgraphs, and show that, in contrast to the low-density type II subgraphs, the highly abundant type I subgraphs cannot exist in isolation but must naturally aggregate into subgraph clusters. The identified topological framework may have important implications for our understanding of the origin and function of subgraphs in all complex networks.
G. Palla, I. Farkas, I. Derenyi, A.-L. Barabási, T. Vicsek
Reverse engineering of linking preferences from network restructuring
Physical Review E 70, 046115 (2004)
Read the abstract
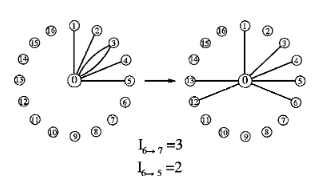
We provide a method to deduce the preferences governing the restructuring dynamics of a network from the observed rewiring of the edges. Our approach is applicable for systems in which the preferences can be formulated in terms of a single-vertex energy function with fskd being the contribution of a node of degree k to the total energy, and the dynamics obeys the detailed balance. The method is first tested by Monte Carlo simulations of restructuring graphs with known energies; then it is used to study variations of real network systems ranging from the coauthorship network of scientific publications to the asset graphs of the New York Stock Exchange. The empirical energies obtained from the restructuring can be described by a universal function fskd,−k ln k, which is consistent with and justifies the validity of the preferential attachment rule proposed for growing networks.
S. Y. Yook, Z. N. Oltvai, A.-L. Barabási
Functional and topological characterization of protein interaction networks
Proteomics 4, 928-942 (2004)
Read the abstract
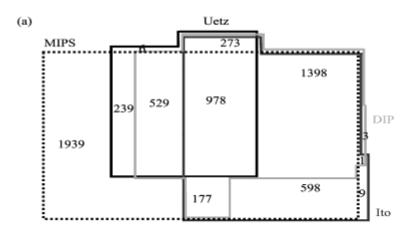
The elucidation of the cell’s large-scale organization is a primary challenge for post-genomic biology, and understanding the structure of protein interaction networks offers an important starting point for such studies. We compare four available databases that approximate the protein interaction network of the yeast, Saccharomyces cerevisiae, aiming to uncover the network’s generic large-scale properties and the impact of the proteins’ function and cellular localization on the network topology. We show how each database supports a scale-free, topology with hierarchical modularity, indicating that these features represent a robust and generic property of the protein interactions network. We also find strong correlations between the network’s structure and the functional role and subcellular localization of its protein constituents, concluding that most functional and/or localization classes appear as relatively segregated subnetworks of the full protein interaction network. The uncovered systematic differences between the four protein interaction databases reflect their relative coverage for different functional and localization classes and provide a guide for their utility in various bioinformatics studies.
R. Dobrin, Q. K. Beg, A.-L. Barabási
Aggregation of topological motifs in the Escherichia coli transcriptional regulatory networks
BMC Bioinformatics 5, 10 (2004)
Read the abstract
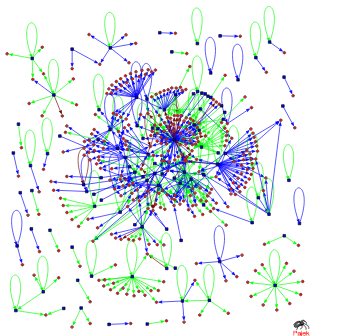
Background: Transcriptional regulation of cellular functions is carried out through a complex network of interactions among transcription factors and the promoter regions of genes and operons regulated by them.To better understand the system-level function of such networks simplification of their architecture was previously achieved by identifying the motifs present in the network, which are small, overrepresented, topologically distinct regulatory interaction patterns (subgraphs). However, the interaction of such motifs with each other, and their form of integration into the full network has not been previously examined. Results: By studying the transcriptional regulatory network of the bacterium, Escherichia coli, we demonstrate that the two previously identified motif types in the network (i.e., feed-forward loops and bi-fan motifs) do not exist in isolation, but rather aggregate into homologous motif clusters that largely overlap with known biological functions. Moreover, these clusters further coalesce into a supercluster, thus establishing distinct topological hierarchies that show global statistical properties similar to the whole network. Targeted removal of motif links disintegrates the network into small, isolated clusters, while random disruptions of equal number of links do not cause such an effect. Conclusion: Individual motifs aggregate into homologous motif clusters and a supercluster forming the backbone of the E. coli transcriptional regulatory network and play a central role in defining its global topological organization.
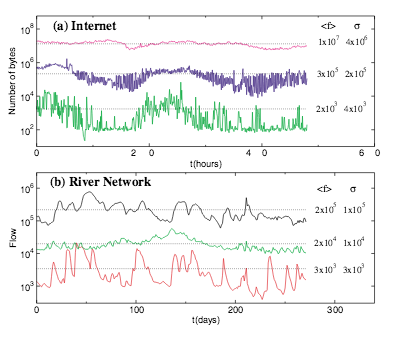
M. A. de Menezes, A.-L. Barabási
Fluctuations in network dynamics
Physical Review Letters 92, 28701 (2004)
Read the abstract
Most complex networks serve as conduits for various dynamical processes, ranging from mass transfer by chemical reactions in the cell to packet transfer on the Internet.We collected data on the time dependent activity of five natural and technological networks, finding that for each the coupling of the flux fluctuations with the total flux on individual nodes obeys a unique scaling law. We show that the observed scaling can explain the competition between the system’s internal collective dynamics and changes in the external environment, allowing us to predict the relevant scaling exponents.
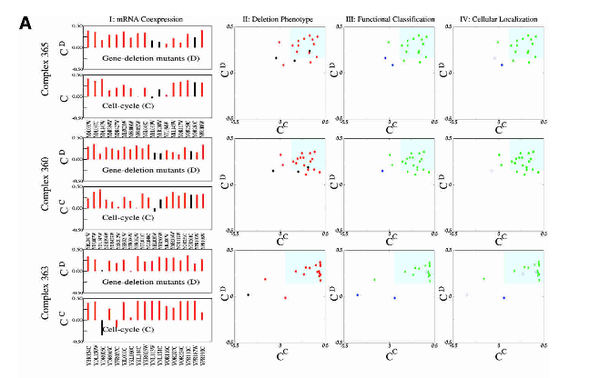
Z. Dezso, Z. N. Oltvai, A.-L. Barabási
Bioinformatics analysis of experimentally determined protein complexes in the yeast Saccharomyces cerevisiae
Genome Research 13, 2450-2454 (2003)
Read the abstract
Many important cellular functions are implemented by protein complexes that act as sophisticated molecular machines of varying size and temporal stability. Here we demonstrate quantitatively that protein complexes in the yeast Saccharomyces cerevisiae are comprised of a core in which subunits are highly coexpressed, display the same deletion phenotype (essential or nonessential), and share identical functional classification and cellular localization. This core is surrounded by a functionally mixed group of proteins, which likely represent short-lived or spurious attachments. The results allow us to define the deletion phenotype and cellular task of most known complexes, and to identify with high confidence the biochemical role of hundreds of proteins with yet unassigned functionality.
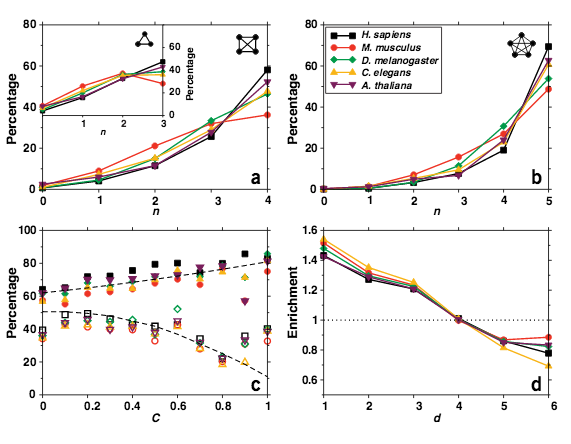
S. Wuchty, Z. N. Oltvai, A.-L. Barabási
Evolutionary conservation of motif constituents in the yeast protein interaction network
Nature Genetics 35, 176-179 (2003)
Read the abstract
Understanding why some cellular components are conserved across species but others evolve rapidly is a key question of modern biology1-3. Here we show that in Saccharomyces cerevisiae, proteins organized in cohesive patterns of interactions are conserved to a substantially higher degree than those that do not participate in such motifs. We find that the conservation of proteins in distinct topological motifs correlates with the interconnectedness and function of that motif and also depends on the structure of the overall interactome topology. These findings indicate that motifs may represent evolutionary conserved topological units of cellular networks molded in accordance with the specific biological function in which they participate.
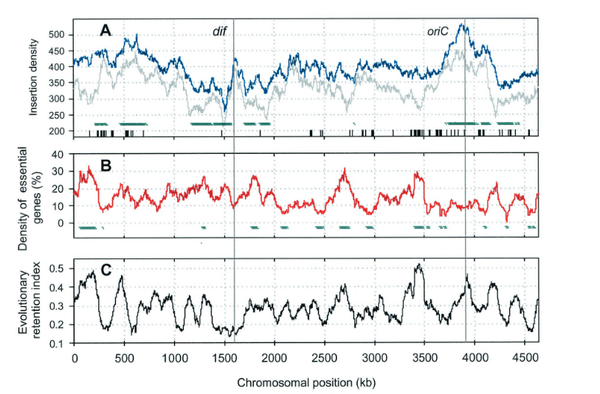
S. Y. Gerdes, M. D. Scholle, J. W. Campbell, G. Balazsi, E. Ravasz, M. D. Daugherty, A. L. Somera, N. C. Kyrpides, I. Anderson, M. S. Gelfand, A. Bhattacharya, V. Kapatral, M. D'Souza, M. V. Baev, Y. Grechkin, F. Mseeh, M. Y. Fonstein, R. Overbeek, A.-L. Barabási, Z. N. Oltvai, A. L. Osterman
Experimental determination and system level analysis of essential genes in Escherichia coli MG1655
Journal of Bacteriology 185, 5673-5684 (2003)
Read the abstract
Defining the gene products that play an essential role in an organism’s functional repertoire is vital to understanding the system level organization of living cells. We used a genetic footprinting technique for a genome-wide assessment of genes required for robust aerobic growth of Escherichia coli in rich media. We identified 620 genes as essential and 3,126 genes as dispensable for growth under these conditions. Functional context analysis of these data allows individual functional assignments to be refined. Evolutionary context analysis demonstrates a ignificant tendency of essential E. coli genes to be preserved throughout the bacterial kingdom. Projection of these data over metabolic subsystems reveals topologic modules with essential and evolutionarily preserved enzymes with reduced capacity for error tolerance.
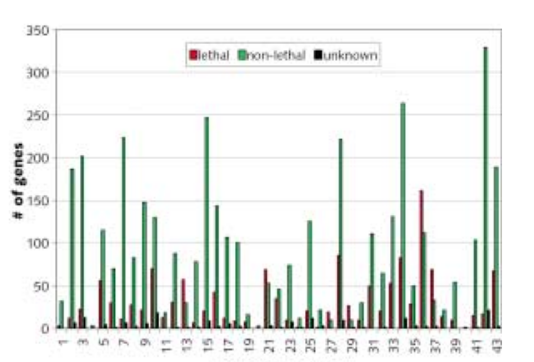
H. Jeong, Z. N. Oltvai, A.-L. Barabási
Prediction of protein essentiality based on genomic data
ComPlexUs 1, 19-28 (2003)
Read the abstract
A major goal of pharmaceutical bioinformatics is to develop computational tools for systematic in silico molecular target identification. Here we demonstrate that in the yeast Saccharomyces cerevisiae the phenotypic effect of single gene deletions simultaneously correlates with fluctuations in mRNA expression profiles, the functional categorization of the gene products, and their connectivity in the yeast’s protein-protein interaction network. Building on these quantitative correlations, we developed a computational method for predicting the phenotypic effect of a given gene’s functional disabling or removal. Our subsequent analyses were in good agreement with the results of systematic gene deletion experiments, allowing us to predict the deletion phenotype of a number of untested yeast genes. The results underscore the utility oflarge genomic databases for in silico systematic drug target identification in the postgenomic era.
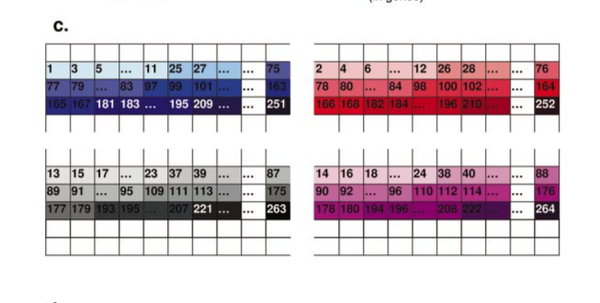
G. Balazsi, K. A. Kay, A.-L. Barabási, Z. N. Oltvai
Spurious spatial periodicity of co-expression in microarray data due to printing design
Nucleic Acids Research 31, 4425-4433 (2003)
Read the abstract
Global transcriptome data is increasingly combined with sophisticated mathematical analyses to extract information about the functional state of a cell. Yet the extent to which the results re¯ect experimental bias at the expense of true biological information remains largely unknown. Here we show that the spatial arrangement of probes on microarrays and the particulars of the printing procedure signi®- cantly affect the log-ratio data of mRNA expression levels measured during the Saccharomyces cerevisiae cell cycle. We present a numerical method that ®lters out these technology-derived contributions from the existing transcriptome data, leading to improved functional predictions. The example presented here underlines the need to routinely search and compensate for inherent experimental bias when analyzing systematically collected,internally consistent biological data sets.

A.-L. Barabási, E. Bonabeau
Scale-Free Networks
Scientific American 288, 50-59 (2003)
Read the abstract
Scientists have recently discovered that various complex systems have an underlying architecture governed by shared organizing principies. This insight has important implications for a host of applications, from drug development to Internet security.
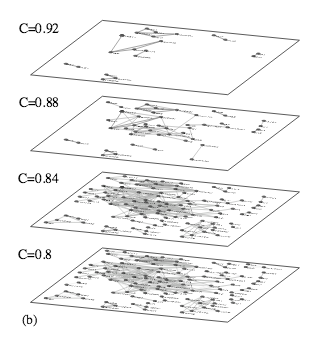
I. Farkas, H. Jeong, T. Vicsek, A.-L. Barabási, Z. N. Oltvai
The topology of the transcription regulatory network in the yeast Saccharomyces cerevisiae
Physica A 318, 601-612 (2003)
Read the abstract
A central goal of postgenomic biology is the elucidation of the regulatory relationships among all cellular constituents that together comprise the ‘genetic network’ of a cell or microorganism. Experimental manipulation of gene activity coupled with the assessment of perturbed transcriptome (i.e., global mRNA expression) patterns represents one approach toward this goal, and may provide a backbone into which other measurements can be later integrated. We use microarray data on 287 single gene deletion Saccharomyces cerevisiae mutant strains to elucidate generic relationships among perturbed transcriptomes. Their comparison with a method that preferentially recognizes distinct expression subpatterns allows us to pair those transcriptomes that share localized similarities. Analyses of the resulting transcriptome similarity network identify a continuum hierarchy among the deleted genes, and in the frequency of local similarities that establishes the links among their reorganized transcriptomes. We also find a combinatorial utilization of shared expression subpatterns within individual links, with increasing quantitative similarity among those that connect transcriptome states induced by the deletion of functionally related gene products. This suggests a distinct hierarchical and combinatorial organization of the S. cerevisiae transcriptional activity, and may represent a pattern that is generic to the transcriptional organization of all eukaryotic organisms.
Z. N. Oltvai, A.-L. Barabási
Life’s complexity pyramid
Science 298, 763-764 (2002)
Read the abstract
Cells and microorganisms have an impressive capacity for adjusting their intracellular machinery in response to changes in their environment, food availability, and developmental state. Add to this an amazing ability to correct internal errors — battling the effects of such mistakes as mutations or misfolded proteins — and we arrive at a major issue of contemporary cell biology: our need to comprehend the staggering complexity, versatility, and robustness of living systems. Although molecular biology offers many spectacular successes, it is clear that the detailed inventory of genes, proteins, and metabolites is not sufficient to understand the cell’s complexity (1). As demonstrated by two papers in this issue—Lee et al. (2) on page 799 and Milo et al. (3) on page 824—viewing the cell as a network of genes and proteins offers a viable strategy for addressing the complexity of living systems.
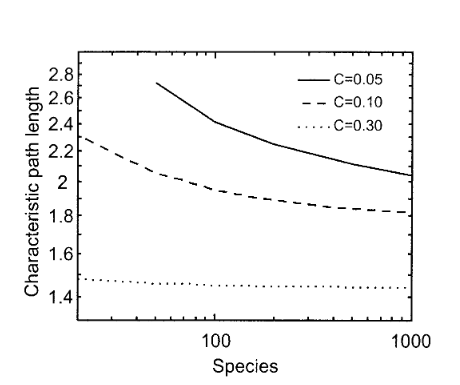
R. J. Williams, N. D. Martinez, E. L. Berlow, J. A. Dunne, A.-L. Barabási
Two degrees of separation in complex food webs
Proceedings of the National Academy of Sciences 99, 12913-12916 (2002)
Read the abstract
Feeding relationships can cause invasions, extirpations, and population fluctuations of a species to dramatically affect other species within a variety of natural habitats. Empirical evidence suggests that such strong effects rarely propagate through food webs more than three links away from the initial perturbation. However, the size of these spheres of potential influence within complex communities is generally unknown. Here, we show for that species within large communities from a variety of aquatic and terrestrial ecosystems are on average two links apart, with >95% of species typically within three links of each other. Species are drawn even closer as network complexity and, more unexpectedly, species richness increase. Our findings are based on seven of the largest and most complex food webs available as well as a food-web model that extends the generality of the empirical results. These results indicate that the dynamics of species within ecosystems may be more highly interconnected and that biodiversity loss and species invasions may affect more species than previously thought.
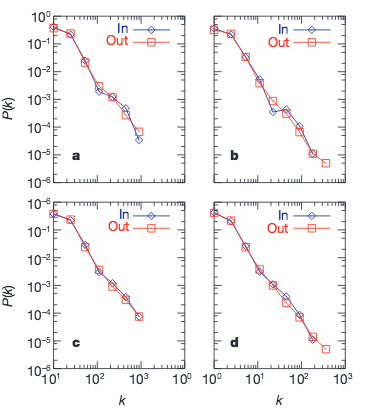
H. Jeong, B. Tombor, R. Albert, Z. N. Oltvai, A.-L. Barabási
The large-scale organization of metabolic networks
Nature 407, 651–655 (2000)
Read the abstract
Here we present a systematic comparative mathematical analysis of the metabolic networks of 43 organisms representing all three domains of life.We show that, despite significant variation in their individual constituents and pathways, these metabolic networks have the same topological scaling properties and show striking similarities to the inherent organization of complex non-biological systems. This may indicate that metabolic organization is not only identical for all living organisms, but also complies with the design principles of robust and error-tolerant scale-free networks, and may represent a common blueprint for the large-scale organization of interactions among all cellular constituents.

J. Podani, Z. N. Oltvai, H. Jeong, B. Tombor, A.-L. Barabási, E. Szathmary
Comparable system-level organization of Archea and Eucaryotes
Nature Genetics 29, 54-56 (2001)
Read the abstract
A central and long-standing issue in evolutionary theory is the origin of the biological variation upon which natural selection acts1. Some hypotheses suggest that evolutionary change represents an adaptation to the surrounding environment within the constraints of an organism’s innate characteristics1–3. Elucidation of the origin and evolutionary relationship of species has been complemented by nucleotide sequence4 and gene content5 analyses, with profound implications for recognizing life’s major domains4. Understanding of evolutionary relationships may be further expanded by comparing systemic higher-level organization among species. Here we employ multivariate analyses to evaluate the biochemical reaction pathways characterizing 43 species. Comparison of the information transfer pathways of Archaea and Eukaryotes indicates a close relationship between these domains. In addition, whereas eukaryotic metabolic enzymes are primarily of bacterial origin6, the pathway-level organization of archaeal and eukaryotic metabolic networks is more closely related. Our analyses therefore suggest that during the symbiotic evolution of eukaryotes, 7–9 incorporation of bacterial metabolic enzymes into the proto-archaeal proteome was constrained by the host’s pre-existing metabolic architecture.
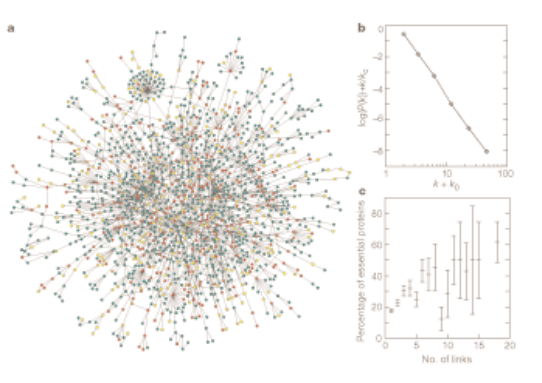
H. Jeong, S. P. Mason, A.-L. Barabási, Z. N. Oltvai
Lethality and centrality in protein networks
Nature 411, 41-42 (2001)
Read the abstract
The most highly connected proteins in the cell are the most important for its survival. Proteins are traditionally identified on the basis of their individual actions as catalysts, signalling molecules, or building blocks in cells and microorganisms. But our post-genomic view is expanding the protein’s role into an element in a network of protein–protein interactions as well, in which it has a contextual or cellular function within functional modules1,2. Here we provide quantitative support for this idea by demonstrating that the phenotypic consequence of a single gene deletion in the yeast Saccharomyces cerevisiae is affected to a large extent by the topological position of its protein product in the complex hierarchical web of molecular interactions.
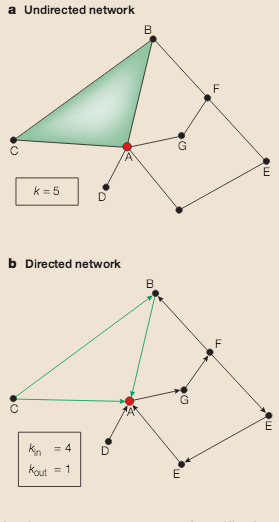
A.-L. Barabási, Z. N. Oltvai
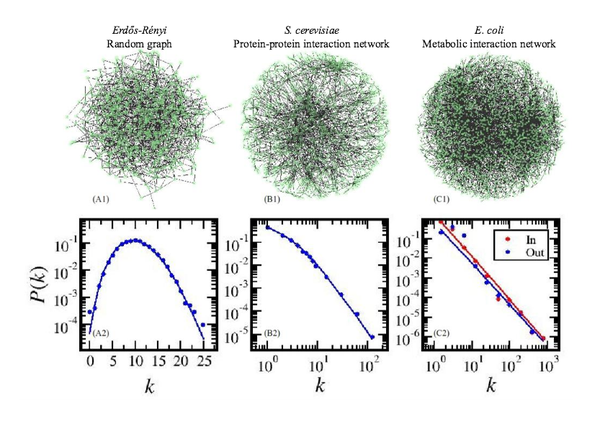
Network biology: understanding the cell’s functional organization
Nature Reviews Genetics 5, 101-113 (2004)
Read the abstract
A key aim of postgenomic biomedical research is to systematically catalogue all molecules and their interactions within a living cell. There is a clear need to understand how these molecules and the interactions between them determine the function of this enormously complex machinery, both in isolation and when surrounded by other cells. Rapid advances in network biology indicate that cellular networks are governed by universal laws and offer a new conceptual framework that could potentially revolutionize our view of biology and disease pathologies in the twenty-first century.
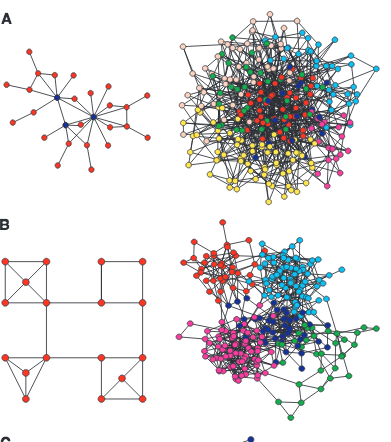
E. Ravasz, A. L. Somera, D. A. Mongru, Z. N. Oltvai, A.-L. Barabási
Hierarchical organization of modularity in metabolic networks
Science 297, 1551-1555 (2002)
Read the abstract
Spatially or chemically isolated functional modules composed of several cellular components and carrying discrete functions are considered fundamental building blocks of cellular organization, but their presence in highly integrated biochemical networks lacks quantitative support. Here, we show that the metabolic networks of 43 distinct organisms are organized into many small, highly connected topologic modules that combine in a hierarchical manner into larger, less cohesive units, with their number and degree of clustering following a power law. Within Escherichia coli, the uncovered hierarchical modularity closely overlaps with known metabolic functions. The identified network architecture may be generic to system-level cellular organization.