Deisy Morselli Gysi, Ítalo do Valle, Marinka Zitnik, Asher Ameli, Xiao Gan, Onur Varol, Susan Dina Ghiassian, J. J. Patten, Robert A. Davey, Joseph Loscalzo, and Albert-László Barabási
Network medicine framework for identifying drug-repurposing opportunities for COVID-19
PNAS May 11, 2021 118 (19) e2025581118
Read the abstract
The COVID-19 pandemic has highlighted the need to quickly and reliably prioritize clinically approved compounds for their potential effectiveness for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infections. Here, we deployed algorithms relying on artificial intelligence, network diffusion, and network proximity, tasking each of them to rank 6,340 drugs for their expected efficacy against SARS-CoV-2. To test the predictions, we used as ground truth 918 drugs experimentally screened in VeroE6 cells, as well as the list of drugs in clinical trials that capture the medical community’s assessment of drugs with potential COVID-19 efficacy. We find that no single predictive algorithm offers consistently reliable outcomes across all datasets and metrics. This outcome prompted us to develop a multimodal technology that fuses the predictions of all algorithms, finding that a consensus among the different predictive methods consistently exceeds the performance of the best individual pipelines. We screened in human cells the top-ranked drugs, obtaining a 62% success rate, in contrast to the 0.8% hit rate of nonguided screenings. Of the six drugs that reduced viral infection, four could be directly repurposed to treat COVID-19, proposing novel treatments for COVID-19. We also found that 76 of the 77 drugs that successfully reduced viral infection do not bind the proteins targeted by SARS-CoV-2, indicating that these network drugs rely on network-based mechanisms that cannot be identified using docking-based strategies. These advances offer a methodological pathway to identify repurposable drugs for future pathogens and neglected diseases underserved by the costs and extended timeline of de novo drug development.

Italo F. do Valle, Harvey G. Roweth, Michael W. Malloy, Sofia Moco, Denis Barron, Elisabeth Battinelli, Joseph Loscalzo & Albert-László Barabási
Network medicine framework shows that proximity of polyphenol targets and disease proteins predicts therapeutic effects of polyphenols
Nature Food volume 2, pages143–155(2021)
Read the abstract
Polyphenols, natural products present in plant-based foods, play a protective role against several complex diseases through their antioxidant activity and by diverse molecular mechanisms. Here we develop a network medicine framework to uncover mechanisms for the effects of polyphenols on health by considering the molecular interactions between polyphenol protein targets and proteins associated with diseases. We find that the protein targets of polyphenols cluster in specific neighbourhoods of the human interactome, whose network proximity to disease proteins is predictive of the molecule’s known therapeutic effects. The methodology recovers known associations, such as the effect of epigallocatechin-3-O-gallate on type 2 diabetes, and predicts that rosmarinic acid has a direct impact on platelet function, representing a novel mechanism through which it could affect cardiovascular health. We experimentally confirm that rosmarinic acid inhibits platelet aggregation and α-granule secretion through inhibition of protein tyrosine phosphorylation, offering direct support for the predicted molecular mechanism. Our framework represents a starting point for mechanistic interpretation of the health effects underlying food-related compounds, allowing us to integrate into a predictive framework knowledge on food metabolism, bioavailability and drug interaction.
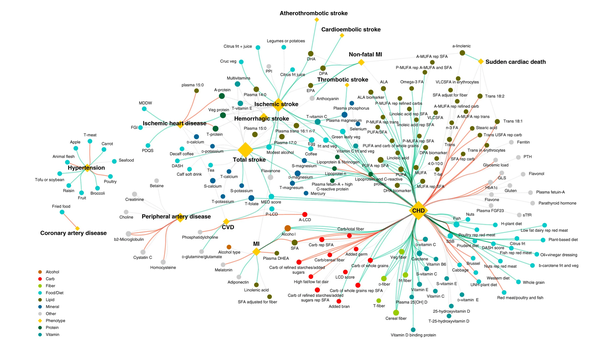
Soodabeh Milanlouei, Giulia Menichetti, Yanping Li, Joseph Loscalzo, Walter C. Willett & Albert-László Barabási
A systematic comprehensive longitudinal evaluation of dietary factors associated with acute myocardial infarction and fatal coronary heart disease
Nature Communications 11, 6074 (2020)
Read the abstract
Environmental factors, and in particular diet, are known to play a key role in the development of Coronary Heart Disease. Many of these factors were unveiled by detailed nutritional epidemiology studies, focusing on the role of a single nutrient or food at a time. Here, we apply an Environment-Wide Association Study approach to Nurses’ Health Study data to explore comprehensively and agnostically the association of 257 nutrients and 117 foods with coronary heart disease risk (acute myocardial infarction and fatal coronary heart disease). After accounting for multiple testing, we identify 16 food items and 37 nutrients that show statistically significant association – while adjusting for potential confounding and control variables such as physical activity, smoking, calorie intake, and medication use – among which 38 associations were validated in Nurses’ Health Study II. Our implementation of Environment-Wide Association Study successfully reproduces prior knowledge of diet-coronary heart disease associations in the epidemiological literature, and helps us detect new associations that were only marginally studied, opening potential avenues for further extensive experimental validation. We also show that Environment-Wide Association Study allows us to identify a bipartite food-nutrient network, highlighting which foods drive the associations of specific nutrients with coronary heart disease risk.
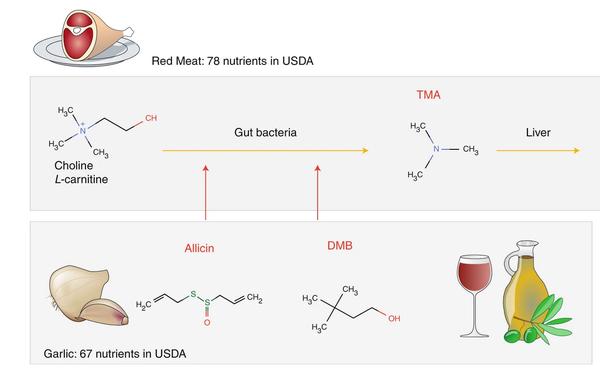
Albert-László Barabási, Giulia Menichetti & Joseph Loscalzo
The unmapped chemical complexity of our diet
Nature Food 1, 33-37 (2019)
Read the abstract
Our understanding of how diet affects health is limited to 150 key nutritional components that are tracked and catalogued by the United States Department of Agriculture and other national databases. Although this knowledge has been transformative for health sciences, helping unveil the role of calories, sugar, fat, vitamins and other nutritional factors in the emergence of common diseases, these nutritional components represent only a small fraction of the more than 26,000 distinct, definable biochemicals present in our food—many of which have documented effects on health but remain unquantified in any systematic fashion across different individual foods. Using new advances such as machine learning, a high-resolution library of these biochemicals could enable the systematic study of the full biochemical spectrum of our diets, opening new avenues for understanding the composition of what we eat, and how it affects health and disease.
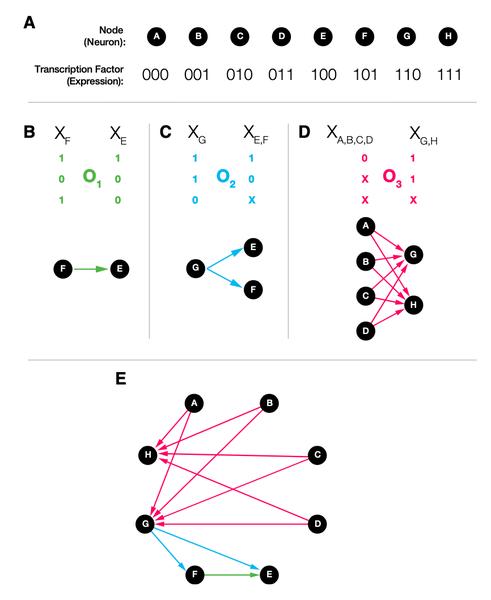
Dániel L. Barabási, Albert-László Barabási
A Genetic Model of the Connectome
Neuron 105, 1-11 Dec 2, 2019
Read the abstract
The connectomes of organisms of the same species show remarkable architectural and often local wiring similarity, raising the question: where and how is neuronal connectivity encoded? Here, we start from the hypothesis that the genetic identity of neurons guides synapse and gap-junction formation and show that such genetically driven wiring predicts the existence of specific biclique motifs in the connectome. We identify a family of large, statistically significant biclique subgraphs in the connectomes of three species and show that within many of the observed bicliques the neurons share statistically significant expression patterns and morphological characteristics, supporting our expectation of common genetic factors that drive the synapse formation within these subgraphs. The proposed connectome model offers a self-consistent framework to link the genetics of an organism to the reproducible architecture of its connectome, offering experimentally falsifiable predictions on the genetic factors that drive the formation of individual neuronal circuits.
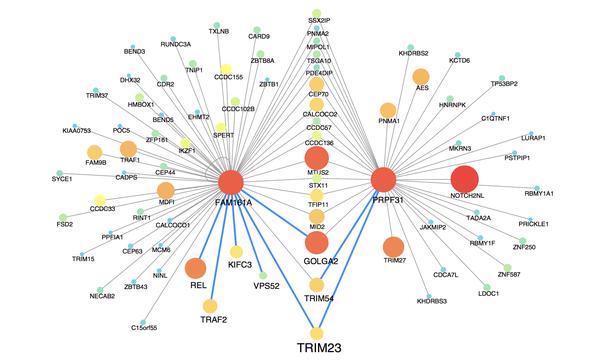
István A. Kovács, Katja Luck, Kerstin Spirohn, Yang Wang, Carl Pollis, Sadie Schlabach, Wenting Bian, Dae-Kyum Kim, Nishka Kishore, Tong Hao, Michael A. Calderwood, Marc Vidal & Albert-László Barabási
Network-based prediction of protein interactions
Nature Communications 10, Article number: 1240 (2019)
Read the abstract
Despite exceptional experimental efforts to map out the human interactome, the continued data incompleteness limits our ability to understand the molecular roots of human disease. Computational tools offer a promising alternative, helping identify biologically significant, yet unmapped protein-protein interactions (PPIs). While link prediction methods connect proteins on the basis of biological or network-based similarity, interacting proteins are not necessarily similar and similar proteins do not necessarily interact. Here, we offer structural and evolutionary evidence that proteins interact not if they are similar to each other, but if one of them is similar to the other’s partners. This approach, that mathematically relies on network paths of length three (L3), significantly outperforms all existing link prediction methods. Given its high accuracy, we show that L3 can offer mechanistic insights into disease mechanisms and can complement future experimental efforts to complete the human interactome.
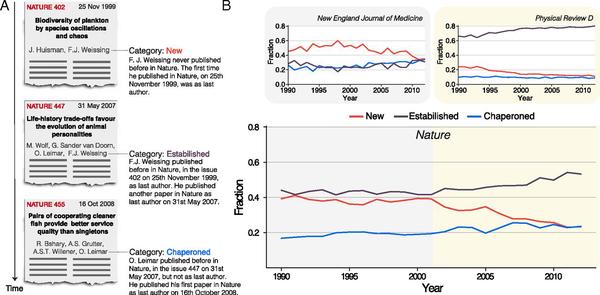
V. Sekara, P. Deville, S. E. Ahnert, A.-L. Barabasi, R. Sinatra, and S. Lehmann
The chaperone effect in scientific publishing
PNAS 115:50, 12603-12607 (2018) https://doi.org/10.1073/pnas.1800471115
Read the abstract
Experience plays a critical role in crafting high-impact scientific work. This is particularly evident in top multidisciplinary journals, where a scientist is unlikely to appear as senior author if he or she has not previously published within the same journal. Here, we develop a quantitative understanding of author order by quantifying this “chaperone effect,” capturing how scientists transition into senior status within a particular publication venue. We illustrate that the chaperone effect has a different magnitude for journals in different branches of science, being more pronounced in medical and biological sciences and weaker in natural sciences. Finally, we show that in the case of high-impact venues, the chaperone effect has significant implications, specifically resulting in a higher average impact relative to papers authored by new principal investigators (PIs). Our findings shed light on the role played by experience in publishing within specific scientific journals, on the paths toward acquiring the necessary experience and expertise, and on the skills required to publish in prestigious venues.

N. Dehmamy, S. Milanlouei, A.-L. Barabasi
A structural transition in physical networks
Nature 563, 676-680 (2018)
Read the abstract
In many physical networks, including neurons in the brain, three-dimensional integrated circuits and underground hyphal networks, the nodes and links are physical objects that cannot intersect or overlap with each other. To take this into account, non-crossing conditions can be imposed to constrain the geometry of networks, which consequently affects how they form, evolve and function. However, these constraints are not included in the theoretical frameworks that are currently used to characterize real networks. Most tools for laying out networks are variants of the force-directed layout algorithm—which assumes dimensionless nodes and links—and are therefore unable to reveal the geometry of densely packed physical networks. Here we develop a modelling framework that accounts for the physical sizes of nodes and links, allowing us to explore how non-crossing conditions affect the geometry of a network. For small link thicknesses, we observe a weakly interacting regime in which link crossings are avoided via local link rearrangements, without altering the overall geometry of the layout compared to the force-directed layout. Once the link thickness exceeds a threshold, a strongly interacting regime emerges in which multiple geometric quantities, such as the total link length and the link curvature, scale with the link thickness. We show that the crossover between the two regimes is driven by the non-crossing condition, which allows us to derive the transition point analytically and show that networks with large numbers of nodes will ultimately exist in the strongly interacting regime. We also find that networks in the weakly interacting regime display a solid-like response to stress, whereas in the strongly interacting regime they behave in a gel-like fashion. Networks in the weakly interacting regime are amenable to 3D printing and so can be used to visualize network geometry, and the strongly interacting regime provides insights into the scaling of the sizes of densely packed mammalian brains.
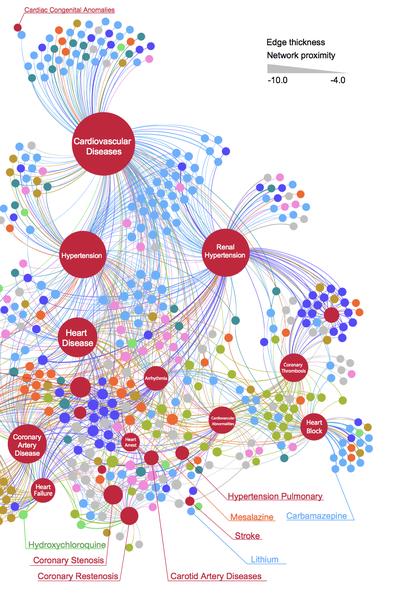
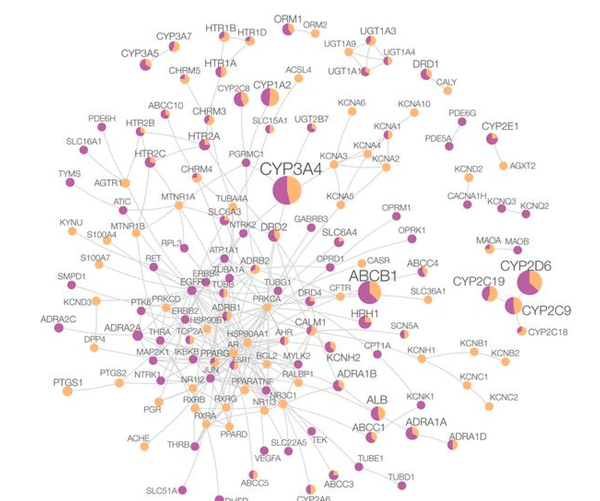
Feixiong Cheng, Rishi J. Desai, Diane E. Handy, Ruisheng Wang, Sebastian Schneeweiss, Albert-László Barabási & Joseph Loscalzo
Network-based approach to prediction and population-based validation of in silico drug repurposing
Nature Communicationsvolume 9, Article number: 2691 (2018)
Read the abstract
Here we identify hundreds of new drug-disease associations for over 900 FDA-approved drugs by quantifying the network proximity of disease genes and drug targets in the human (protein–protein) interactome. We select four network-predicted associations to test their causal relationship using large healthcare databases with over 220 million patients and state-of-the-art pharmacoepidemiologic analyses. Using propensity score matching, two of four network-based predictions are validated in patient-level data: carbamazepine is associated with an increased risk of coronary artery disease (CAD) [hazard ratio (HR) 1.56, 95% confidence interval (CI) 1.12–2.18], and hydroxychloroquine is associated with a decreased risk of CAD (HR 0.76, 95% CI 0.59–0.97). In vitro experiments show that hydroxychloroquine attenuates pro-inflammatory cytokine-mediated activation in human aortic endothelial cells, supporting mechanistically its potential beneficial effect in CAD. In summary, we demonstrate that a unique integration of protein-protein interaction network proximity and large-scale patient-level longitudinal data complemented by mechanistic in vitro studies can facilitate drug repurposing.
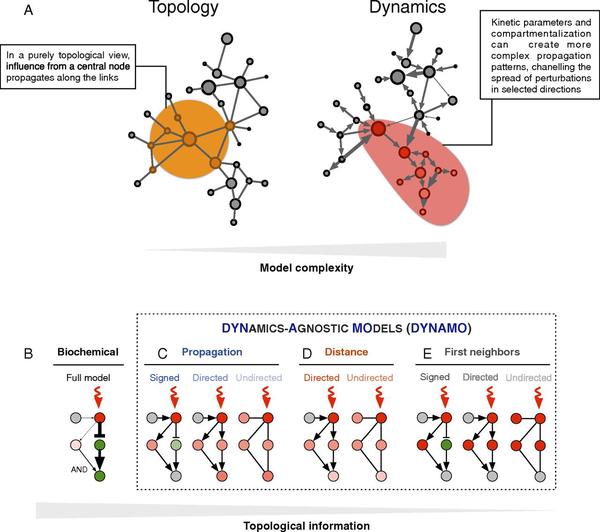
M. Santolini, A.-L. Barabasi
Predicting Perturbation Patterns from the Topology of Biological Networks
PNAS | vol. 115 | no. 27 | E6375–E6383
Read the abstract
High-throughput technologies, offering an unprecedented wealth of quantitative data underlying the makeup of living systems, are changing biology. Notably, the systematic mapping of the relationships between biochemical entities has fueled the rapid development of network biology, offering a suitable framework to describe disease phenotypes and predict potential drug targets. However, our ability to develop accurate dynamical models remains limited, due in part to the limited knowledge of the kinetic parameters underlying these interactions. Here, we explore the degree to which we can make reasonably accurate predictions in the absence of the kinetic parameters. We find that simple dynamically agnostic models are sufficient to recover the strength and sign of the biochemical perturbation patterns observed in 87 biological models for which the underlying kinetics are known. Surprisingly, a simple distance-based model achieves 65% accuracy. We show that this predictive power is robust to topological and kinetic parameter perturbations, and we identify key network properties that can increase up to 80% the recovery rate of the true perturbation patterns. We validate our approach using experimental data on the chemotactic pathway in bacteria, finding that a network model of perturbation spreading predicts with ∼80% accuracy the directionality of gene expression and phenotype changes in knock-out and overproduction experiments. These findings show that the steady advances in mapping out the topology of biochemical interaction networks opens avenues for accurate perturbation spread modeling, with direct implications for medicine and drug development.
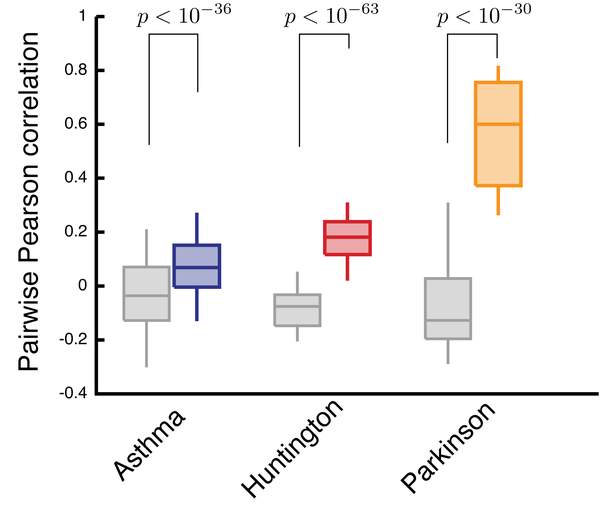
J. Menche, E. Guney, A. Sharma, P. J. Branigan, M. J. Loza, F. Baribaud, R. Dobrin, A.-L. Barabasi
Integrating personalized gene expression profiles into predictive disease-associated gene pools
Systems Biology and Applications 3:10 (2017)
Read the abstract
Gene expression data are routinely used to identify genes that on average exhibit different expression levels between a case and a control group. Yet, very few of such differentially expressed genes are detectably perturbed in individual patients. Here, we develop a framework to construct personalized perturbation profiles for individual subjects, identifying the set of genes that are significantly perturbed in each individual. This allows us to characterize the heterogeneity of the molecular manifestations of complex diseases by quantifying the expression-level similarities and differences among patients with the same phenotype. We show that despite the high heterogeneity of the individual perturbation profiles, patients with asthma, Parkinson and Huntington’s disease share a broadpool of sporadically disease-associated genes, and that individuals with statistically signi ficant overlap with this pool have a 80–100% chance of being diagnosed with the disease. The developed framework opens up the possibility to apply gene expression data in the context of precision medicine, with important implications for biomarker identifi cation, drug development, diagnosis and treatment.
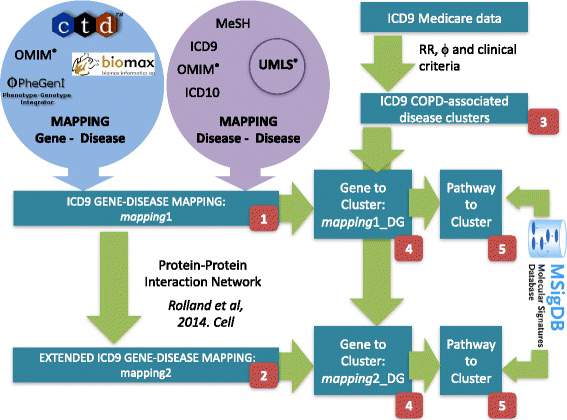
D. Gomez-Cabrero, J. Menche, C. Vargas, I. Cano, D. Maier, A.-L. Barabasi, J. Tegner, J. Roca (Synergy-COPD Consortia)
From Comorbidities of Chronic Obstructive Pulmonary Disease to Identification of Shared Molecular Mechanisms by Data Integration
BMC Bioinformatics 17: 1291 (2016)
Read the abstract
Background
Deep mining of healthcare data has provided maps of comorbidity relationships between diseases. In parallel, integrative multi-omics investigations have generated high-resolution molecular maps of putative relevance for understanding disease initiation and progression. Yet, it is unclear how to advance an observation of comorbidity relations (one disease to others) to a molecular understanding of the driver processes and associated biomarkers.
Results
Since Chronic Obstructive Pulmonary disease (COPD) has emerged as a central hub in temporal comorbidity networks, we developed a systematic integrative data-driven framework to identify shared disease-associated genes and pathways, as a proxy for the underlying generative mechanisms inducing comorbidity. We integrated records from approximately 13 M patients from the Medicare database with disease-gene maps that we derived from several resources including a semantic-derived knowledge-base. Using rank-based statistics we not only recovered known comorbidities but also discovered a novel association between COPD and digestive diseases. Furthermore, our analysis provides the first set of COPD co-morbidity candidate biomarkers, including IL15, TNF and JUP, and characterizes their association to aging and life-style conditions, such as smoking and physical activity.
Conclusions
The developed framework provides novel insights in COPD and especially COPD co-morbidity associated mechanisms. The methodology could be used to discover and decipher the molecular underpinning of other comorbidity relationships and furthermore, allow the identification of candidate co-morbidity biomarkers.
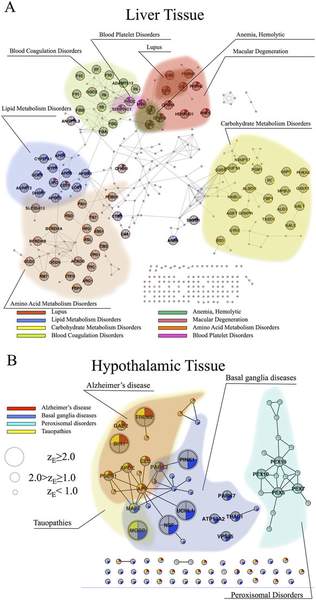
M. Kitsak, A. Sharma, J. Menche, E. Guney, S. D. Ghiassian, J. Loscalzo, A.-L. Barabasi
Tissue Specificity of Human Disease Module
Scientific Reports 6: 35241 (2016)
Read the abstract
Genes carrying mutations associated with genetic diseases are present in all human cells; yet, clinical manifestations of genetic diseases are usually highly tissue-specific. Although some disease genes are expressed only in selected tissues, the expression patterns of disease genes alone cannot explain the observed tissue specificity of human diseases. Here we hypothesize that for a disease to manifest itself in a particular tissue, a whole functional subnetwork of genes (disease module) needs to be expressed in that tissue. Driven by this hypothesis, we conducted a systematic study of the expression patterns of disease genes within the human interactome. We find that genes expressed in a specific tissue tend to be localized in the same neighborhood of the interactome. By contrast, genes expressed in different tissues are segregated in distinct network neighborhoods. Most important, we show that it is the integrity and the completeness of the expression of the disease module that determines disease manifestation in selected tissues. This approach allows us to construct a disease-tissue network that confirms known and predicts unexpected disease-tissue associations.

G. Basler, Z. Nikoloski, A. Larhlimi, A.-L. Barabasi, and Y.-Y. Liu
Control of Fluxes in Metabolic Networks
Genome Research 7: 26, 956-968 (2016)
Read the abstract
Understanding the control of large-scale metabolic networks is central to biology and medicine. However, existing approaches either require specifying a cellular objective or can only be used for small networks. We introduce new coupling types describing the relations between reaction activities, and develop an efficient computational framework, which does not require any cellular objective for systematic studies of large-scale metabolism. We identify the driver reactions facilitating control of 23 metabolic networks from all kingdoms of life. We find that unicellular organisms require a smaller degree of control than multicellular organisms. Driver reactions are under complex cellular regulation in Escherichia coli, indicating their preeminent role in facilitating cellular control. In human cancer cells, driver reactions play pivotal roles in malignancy and represent potential therapeutic targets. The developed framework helps us gain insights into regulatory principles of diseases and facilitates design of engineering strategies at the interface of gene regulation, signaling, and metabolism.
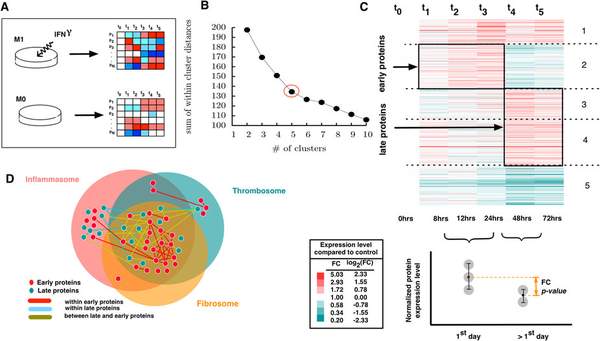
S. D. Ghiassian, J. Menche, D. I. Chasman, F. Giulianini, R. Wang, P. Ricchiuto, M. Aikawa, H. Iwata, C. Muller, T. Zeller, A. Sharma, P. Wild, K. Lackner, S. Singh, P. M. Ridker, S. Blankenberg, A.-L. Barabasi, J. Loscalzo
Endophenotype Network Models: Common Core of Complex Diseases
Scientific Reports 6: 27414, 1-13 (2016)
Read the abstract
Historically, human diseases have been differentiated and categorized based on the organ system in which they primarily manifest. Recently, an alternative view is emerging that emphasizes that different diseases often have common underlying mechanisms and shared intermediate pathophenotypes, or endo(pheno)types. Within this framework, a specific disease’s expression is a consequence of the interplay between the relevant endophenotypes and their local, organ-based environment. Important examples of such endophenotypes are inflammation, fibrosis, and thrombosis and their essential roles in many developing diseases. In this study, we construct endophenotype network models and explore their relation to different diseases in general and to cardiovascular diseases in particular. We identify the local neighborhoods (module) within the interconnected map of molecular components, i.e., the subnetworks of the human interactome that represent the inflammasome, thrombosome, and fibrosome. We find that these neighborhoods are highly overlapping and significantly enriched with disease-associated genes. In particular they are also enriched with differentially expressed genes linked to cardiovascular disease (risk). Finally, using proteomic data, we explore how macrophage activation contributes to our understanding of inflammatory processes and responses. The results of our analysis show that inflammatory responses initiate from within the cross-talk of the three identified endophenotypic modules.
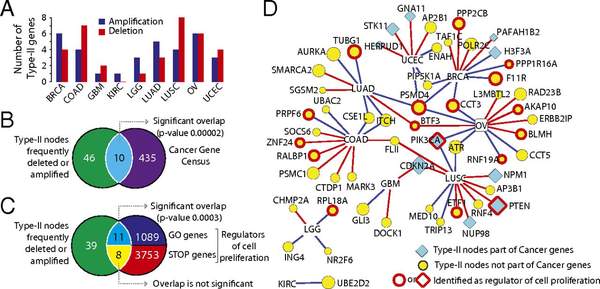
A. Vinayagama, T.E. Gibsonb, H.-J. Lee, B. Yilmazeld, C. Roeseld, Y. Hua, Y. Kwona, A. Sharma, Y.-Y. Liu, N. Perrimona, A.-L. Barabasi
Controllability Analysis of the Directed Human Protein Interaction Network Identifies Disease Genes and Drug Targets
Proceedings of the National Academy of Sciences 10.1073/pnas.1603992113, 1-6 (2016)
Read the abstract
The protein-protein interaction (PPI) network is crucial for cellular information processing and decision-making. With suitable inputs, PPI networks drive the cells to diverse functional outcomes such as cell proliferation or cell death. Here, we characterize the structural controllability of a large directed human PPI network comprising 6,339 proteins and 34,813 interactions. This network allows us to classify proteins as "indispensable," "neutral," or "dispensable," which correlates to increasing, no effect, or decreasing the number of driver nodes in the network upon removal of that protein. We find that 21% of the proteins in the PPI network are indispensable. Interestingly, these indispensable proteins are the primary targets of disease-causing mutations, human viruses, and drugs, suggesting that altering a networks control property is critical for the transition between healthy and disease states. Furthermore, analyzing copy number alterations data from 1,547 cancer patients reveals that 56 genes that are frequently amplified or deleted in nine different cancers are indispensable. Among the 56 genes, 46 of them have not been previously associated with cancer. This suggests that controllability analysis is very useful in identifying novel disease genes and potential drug targets.
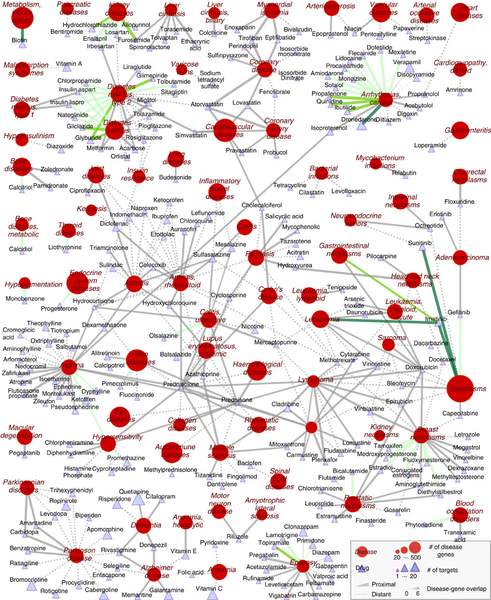
E. Guney, J. Menche, M. Vidal, A.-L. Barábasi
Network-based in silico drug efficacy screening
Nature Communications 7:10331, 1-13 (2016)
Read the abstract
The increasing cost of drug development together with a significant drop in the number of
new drug approvals raises the need for innovative approaches for target identification
and efficacy prediction. Here, we take advantage of our increasing understanding of the
network-based origins of diseases to introduce a drug-disease proximity measure that
quantifies the interplay between drugs targets and diseases. By correcting for the known
biases of the interactome, proximity helps us uncover the therapeutic effect of drugs, as well
as to distinguish palliative from effective treatments. Our analysis of 238 drugs used in 78
diseases indicates that the therapeutic effect of drugs is localized in a small network
neighborhood of the disease genes and highlights efficacy issues for drugs used in Parkinson
and several inflammatory disorders. Finally, network-based proximity allows us to predict
novel drug-disease associations that offer unprecedented opportunities for drug repurposing
and the detection of adverse effects.
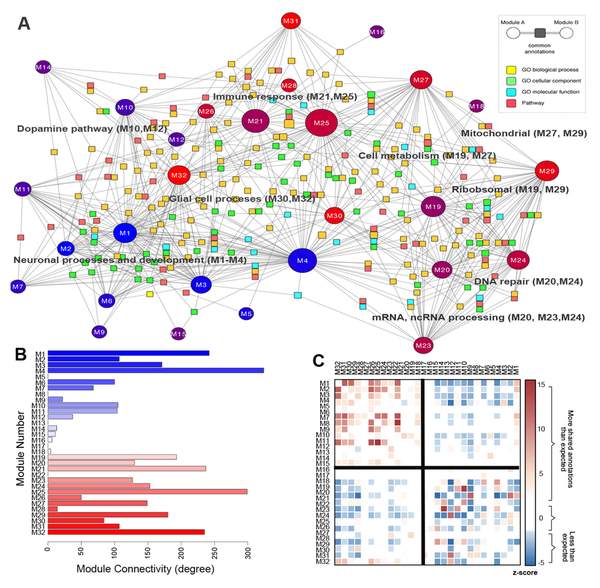
M. Hawrylycz, J. A. Miller, V. Menon, D. Feng, T. Dolbeare, A. L. Guillozet-Bongaarts, A. G. Jegga, B. J. Aronow, C.-K. Lee, A. Bernard, M. F. Glasser, D. L. Dierker, J. Menche, A. Szafer, F. Collman, P. Grange, K. A. Berman, S. Mihalas, Z. Yao, L. Stewart, A.-L. Barabási, J. Schulkin, J. Phillips, L. Ng, C. Dang, D. R. Haynor, A. Jones, D. C. Van Essen, C. Koch, D. Lein
Canonical genetic signatures of the adult human brain
Nature Neuroscience 4171, 1-15 (2015)
Read the abstract
The structure and function of the human brain are highly stereotyped, implying a conserved molecular program responsible for its development, cellular structure and function. We applied a correlation-based metric called differential stability to assess reproducibility of gene expression patterning across 132 structures in six individual brains, revealing mesoscale genetic organization. The genes with the highest differential stability are highly biologically relevant, with enrichment for brain-related annotations, disease associations, drug targets and literature citations. Using genes with high differential stability, we identified 32 anatomically diverse and reproducible gene expression signatures, which represent distinct cell types, intracellular components and/or associations with neurodevelopmental and neurodegenerative disorders. Genes in neuron-associated compared to non-neuronal networks showed higher preservation between human and mouse; however, many diversely patterned genes displayed marked shifts in regulation between species. Finally, highly consistent transcriptional architecture in neocortex is correlated with resting state functional connectivity, suggesting a link between conserved gene expression and functionally relevant circuitry.
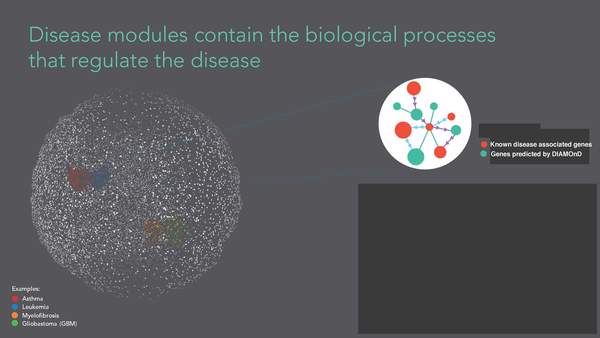
S. D. Ghiassian, J. Menche, A.-L. Barabási
A DIseAse MOdule Detection (DIAMOnD) Algorithm Derived from a Systematic Analysis of Connectivity Patterns of Disease Proteins in the Human Interactome
PLOS Computational Biology pcbi.1004120, 1-21 (2015)
Read the abstract
The observation that disease associated proteins often interact with each other has fueled the development of network-based approaches to elucidate the molecular mechanisms of human disease. Such approaches build on the assumption that protein interaction networks can be viewed as maps in which diseases can be identified with localized perturbation within a certain neighborhood. The identification of these neighborhoods, or disease modules, is therefore a prerequisite of a detailed investigation of a particular pathophenotype. While numerous heuristic methods exist that successfully pinpoint disease associated modules, the basic underlying connectivity patterns remain largely unexplored. In this work we aim to fill this gap by analyzing the network properties of a comprehensive corpus of 70 complex diseases. We find that disease associated proteins do not reside within locally dense communities and instead identify connectivity significance as the most predictive quantity. This quantity inspires the design of a novel Disease Module Detection (DIAMOnD) algorithm to identify the full disease module around a set of known disease proteins. We study the performance of the algorithm using well-controlled synthetic data and systematically validate the identified neighborhoods for a large corpus of diseases.
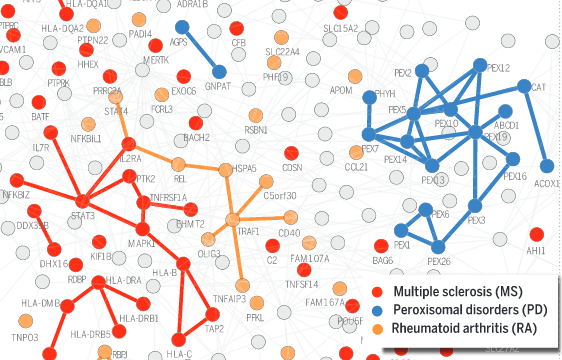
J. Menche, A. Sharma, M. Kitsak, D. Ghiassian, M. Vidal, J. Loscazlo, A.-L. Barabasi
Uncovering disease-disease relationships through the incomplete interactome
Science 347:6224, 1257601-1 (2015)
Read the abstract
According to the disease module hypothesis, the cellular components associated with a disease segregate in the same neighborhood of the human interactome, the map of biologically relevant molecular interactions. Yet, given the incompleteness of the interactome and the limited knowledge of disease-associated genes, it is not obvious if the available data have sufficient coverage to map out modules associated with each disease. Here we derive mathematical conditions for the identifiability of disease modules and show that the network-based location of each disease module determines its pathobiological relationship to other diseases. For example, diseases with overlapping network modules show significant coexpression patterns, symptom similarity, and comorbidity, whereas diseases residing in separated network neighborhoods are phenotypically distinct. These tools represent an interactome-based platform to predict molecular commonalities between phenotypically related diseases, even if they do not share primary disease genes.
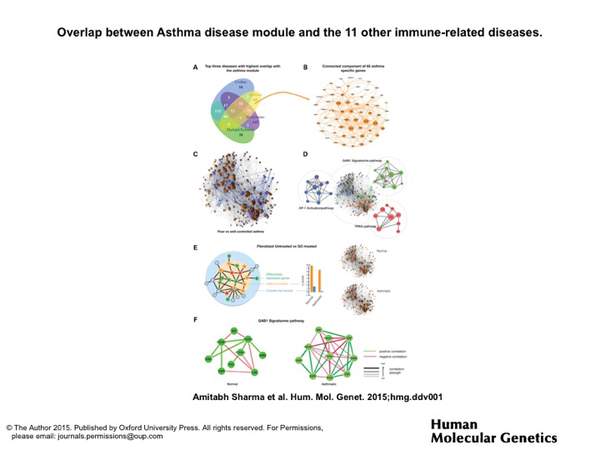
A. Sharma, J. Menche, C. C. Huang, T. Ort, X. Zhou, M. Kitsak, N. Sahni, D. Thibault, L. Voung, F. Guo, S. D. Ghiassian, N. Gulbahce, F. Baribaud, J. Tocker, R. Dobrin, E. Barnathan, H. Liu, R. A. Panettieri Jr., K. G. Tantisira, W. Qiu, B. A. Raby, E. K. Silverman, M. Vidal, S. T. Weiss, and A.-L. Barabási
A disease module in the interactome explains disease heterogeneity, drug response and captures novel pathways and genes in asthma
Human Molecular Genetics 101093, 1-16 (2015)
Read the abstract
Recent advances in genetics have spurred rapid progress towards the systematic identification of genes involved in complex diseases. Still, the detailed understanding of the molecular and physiological mechanisms through which these genes affect disease phenotypes remains a major challenge. Here, we identify the asthma disease module, i.e. the local neighborhood of the interactome whose perturbation is associated with asthma, and validate it for functional and pathophysiological relevance, using both computational and experimental approaches. We find that the asthma disease module is enriched with modest GWAS P-values against the background of random variation, and with differentially expressed genes from normal and asthmatic fibroblast cells treated with an asthma-specific drug. The asthma module also contains immune response mechanisms that are shared with other immune-related disease modules. Further, using diverse omics (genomics,gene-expression, drug response) data,we identify the GAB1 signaling pathway as an important novel modulator in asthma. The wiring diagram of the uncovered asthma module suggests a relatively close link between GAB1 and glucocorticoids (GCs), which we experimentally validate, observing an increase in the level of GAB1 after GC treatment in BEAS-2B bronchial epithelial cells. The siRNA knockdown of GAB1 in the BEAS-2B cell line resulted in a decrease in the NFkB level, suggesting a novel regulatory path of the pro-inflammatory factor NFkB by GAB1 in asthma.
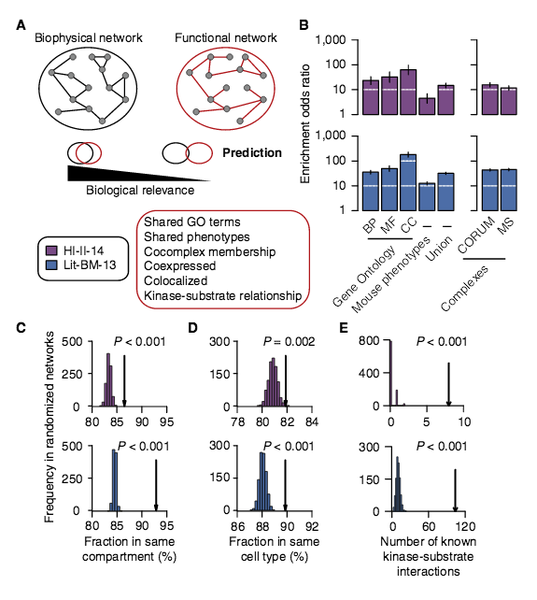
T. Rolland, M. Tasan, , B. Charloteaux, S. J. Pevzner,, Q. Zhong, N. Sahni, S. Yi,, I. Lemmens, C. Fontanillo,, R. Mosca, A. Kamburov, , S. D. Ghiassian, X. Yang,, L. Ghamsari, D. Balcha,, B. E. Begg, P. Braun, M. Brehm, M. P. Froly, A.-R. Carvunis, D, Convery-Zupan, R. Carominas,, J. Coulombe-Huntington, , E. Dann, M. Dreze, A. Dricot,, C. Fan, E. Franzosa, F. Gebrea, B. J. Gutierrez, M. F. Hardy,, M. Jin, S. Kang, R. Kiros, G. , Lin, K. Luck, A. MacWilliams,, J. Menche, R R. Murray, A., Palagi, M. M. Poulin, X. , Rambout, J. Rasla, P. Reichert, V. Romero, E. Ruyssinck, J. M., Sahalie, plus 20 more co-autho
A proteome-scale map of the human interactome network
Cell 159:5, 1212-1226 (2014)
Read the abstract
Just as reference genome sequences revolutionized human genetics, reference maps of interactome networks will be critical to fully understand genotype-phenotype relationships. Here, we describe a systematic map of ∼14,000 high-quality human binary protein-protein interactions. At equal quality, this map is ∼30% larger than what is available from small-scale studies published in the literature in the last few decades. While currently available information is highly biased and only covers a relatively small portion of the proteome, our systematic map appears strikingly more homogeneous, revealing a “broader” human interactome network than currently appreciated. The map also uncovers significant interconnectivity between known and candidate cancer gene products, providing unbiased evidence for an expanded functional cancer landscape, while demonstrating how high-quality interactome models will help “connect the dots” of the genomic revolution.
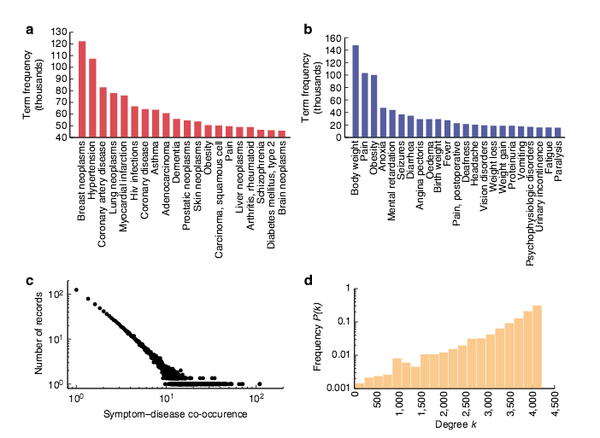
X. Z. Zhou, J. Menche, A.-L. Barabási, A. Sharma
Human symptoms–disease network
Nature Communications 5:4212, 1-10 (2014)
Read the abstract
In the post-genomic era, the elucidation of the relationship between the molecular origins of diseases and their resulting phenotypes is a crucial task for medical research. Here, we use a large-scale biomedical literature database to construct a symptom-based human disease network and investigate the connection between clinical manifestations of diseases and their underlying molecular interactions. We find that the symptom-based similarity of two diseases correlates strongly with the number of shared genetic associations and the extent to which their associated proteins interact. Moreover, the diversity of the clinical manifestations of a disease can be related to the connectivity patterns of the underlying protein interaction network. The comprehensive, high-quality map of disease–symptom relations can further be used as a resource helping to address important questions in the field of systems medicine, for example, the identification of unexpected associations between diseases, disease etiology research or drug design.
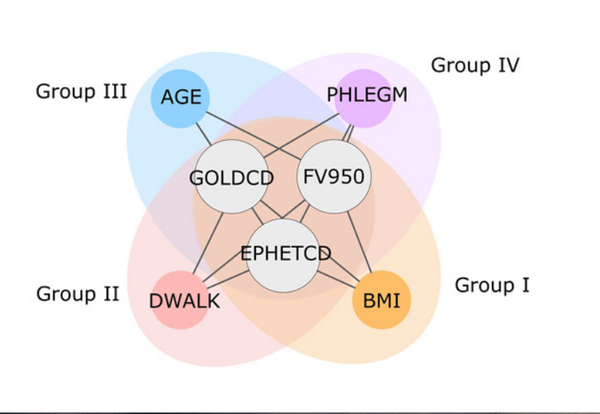
J. Mench, A. Sharma, M. H. Cho, R. J. Mayer, S. I. Rennard, B. Celli, B. E. Miller, N. Locantore, R. Tal-Singer, S. Ghosh, C. Larminie, G. Bradley, J. H. Riley, A. Agusti, E. K. Silverman, A.-L. Barabási
A diVIsive Shuffling Approach (VIStA) for gene expression analysis to identify subtypes in Chronic Obstructive Pulmonary Disease
BMC Systems Biology 8, 1-13 (2014)
Read the abstract
Background: An important step toward understanding the biological mechanisms underlying a complex disease is a refined understanding of its clinical heterogeneity. Relating clinical and molecular differences may allow us to define more specific subtypes of patients that respond differently to therapeutic interventions. Results: We developed a novel unbiased method called diVIsive Shuffling Approach (VIStA) that identifies subgroups of patients by maximizing the difference in their gene expression patterns. We tested our algorithm on 140 subjects with Chronic Obstructive Pulmonary Disease (COPD) and found four distinct, biologically and clinically meaningful combinations of clinical characteristics that are associated with large gene expression differences. The dominant characteristic in these combinations was the severity of airflow limitation. Other frequently identified measures included emphysema, fibrinogen levels, phlegm, BMI and age. A pathway analysis of the differentially expressed genes in the identified subtypes suggests that VIStA is capable of capturing specific molecular signatures within in each group. Conclusions: The introduced methodology allowed us to identify combinations of clinical characteristics that correspond to clear gene expression differences. The resulting subtypes for COPD contribute to a better understanding of its heterogeneity.
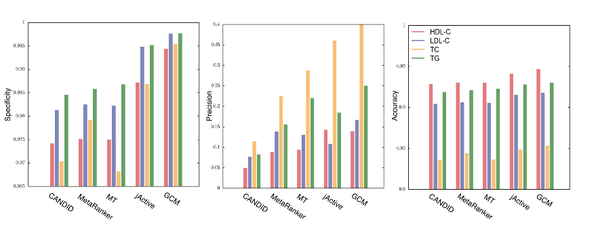
A. Sharma, N. Gulbahce, S. J. Pevzner, J. Menche, C. Ladenvall, L. Folkdersen, P. Eriksson, M. Orho-Melander, A.-L. Barabási
Network-based analysis of genome wide association data provides novel candidate genes for lipid and lipoprotein traits
Molecular & Cellular Proteomics 12, 3398-3408 (2013)
Read the abstract
Genome wide association studies (GWAS) identify susceptibility loci for complex traits, but do not identify particular genes of interest. Integration of functional and network information may help in overcoming this limitation and identifying new susceptibility loci. Using GWAS and comorbidity data, we present a network-based approach to predict candidate genes for lipid and lipoprotein traits. We apply a prediction pipeline incorporating interactome, co-expression, and comorbidity data to Global Lipids Genetics Consortium (GLGC) GWAS for four traits of interest, identifying phenotypically coherent modules. These modules provide insights regarding gene involvement in complex phenotypes with multiple susceptibility alleles and low effect sizes. To experimentally test our predictions, we selected four candidate genes and genotyped representative SNPs in the Malmö Diet and Cancer Cardiovascular Cohort. We found significant associations with LDL-C and total-cholesterol levels for a synonymous SNP (rs234706) in the cystathionine beta-synthase (CBS) gene (p = 1 × 10−5 and adjusted-p = 0.013, respectively). Further, liver samples taken from 206 patients revealed that patients with the minor allele of rs234706 had significant dysregulation of CBS (p = 0.04). Despite the known biological role of CBS in lipid metabolism, SNPs within the locus have not yet been identified in GWAS of lipoprotein traits. Thus, the GWAS-based Comorbidity Module (GCM) approach identifies candidate genes missed by GWAS studies, serving as a broadly applicable tool for the investigation of other complex disease phenotypes.
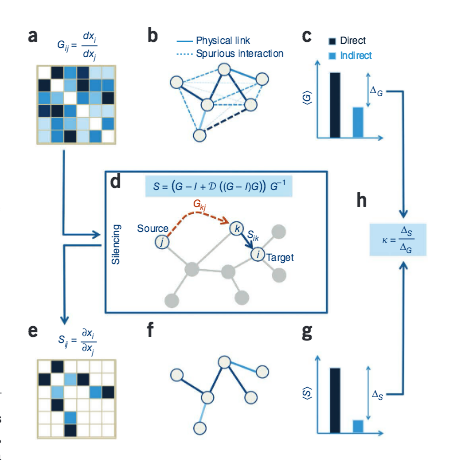
B. Barzel, A.-L. Barabási
Network link prediction by global silencing of indirect correlations
Nature Biotechnology 31: Num 8, 1-8 (2013)
Read the abstract
Predictions of physical and functional links between cellular components are often based on correlations between experimental measurements, such as gene expression. However, correlations are affected by both direct and indirect paths, confounding our ability to identify true pairwise interactions. Here we exploit the fundamental properties of dynamical correlations in networks to develop a method to silence indirect effects. The method receives as input the observed correlations between node pairs and uses a matrix transformation to turn the correlation matrix into a highly discriminative silenced matrix, which enhances only the terms associated with direct causal links. Against empirical data for Escherichia coli regulatory interactions, the method enhanced the discriminative power of the correlations by twofold, yielding >50% predictive improvement over traditional correlation measures and 6% over mutual information. Overall this silencing method will help translate the abundant correlation data into insights about a system's interactions, with applications ranging from link prediction to inferring the dynamical mechanisms governing biological networks.
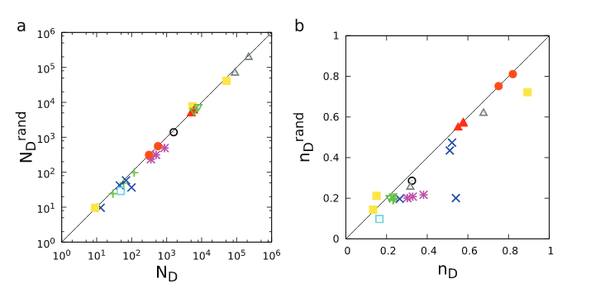
M. Pósfai, Y.-Y. Liu, J.-J. Slotine, A.-L. Barabási
Effect of correlations on network controllability
Scientific Reports 3:1067, 1-7 (2013)
Read the abstract
A dynamical system is controllable if by imposing appropriate external signals on a subset of its nodes, it can be driven from any initial state to any desired state in finite time. Here we study the impact of various network characteristics on the minimal number of driver nodes required to control a network. We find that clustering and modularity have no discernible impact, but the symmetries of the underlying matching problem can produce linear, quadratic or no dependence on degree correlation coefficients, depending on the nature of the underlying correlations. The results are supported by numerical simulations and help narrow the observed gap between the predicted and the observed number of driver nodes in real networks.
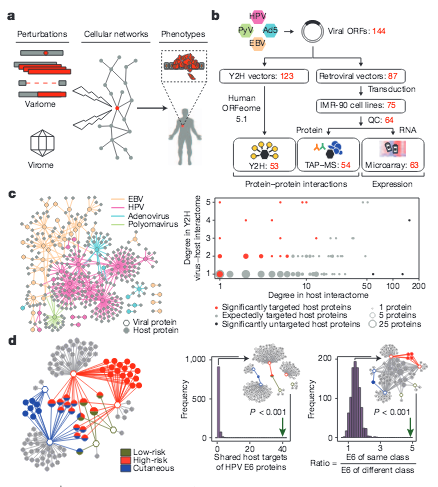
O. Rozenblatt-Rosen, R. C. Deo, M. Padi, G. Adelmant, T. Rolland, M. Grace, A. Dricot, M. Askenazi, M. Tavares, S. J. Pevzner, F. Abderazzaq, D. Byrdsong, A.-R. Carvunis, A. A. Chen, J. Cheng, M. Correll, M. Durate, C. Fan, M. C. Feltkamp, S. B. Ficarro, R. Franchi, B. K. Garg, N. Gulbahce, T. Hao, A. M. Holthaus, R. James, A. Korkhin, L. Litovchick, J. C. Mar, T. R. Pak, S. Rabello, R. Rubio, Y. Shen, S. Singh, J. M. Spangle, M. Tasan, S. Wanamakter, J. T. Webber, J. Roecklein-Canfield,, E. Johannsen, A.-L. Barabasi,, R. Beroukhim, E. Kieff,, M. E. Cusick, D. E. Hill,, K. Munger, J. A. Marto,, J. Quackenbush, F. P. Roth,, J. A. DeCaprio, M. Vidal
Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins
Nature 487, 491-495 (2012)
Read the abstract
Genotypic differences greatly influence susceptibility and resistance to disease. Understanding genotype–phenotype relationships requires that phenotypes be viewed as manifestations of network properties, rather than simply as the result of individual genomic variations. Genome sequencing efforts have identified numerous germline mutations, and large numbers of somatic genomic alterations, associated with a predisposition to cancer. However, it remains difficult to distinguish background, or ‘passenger’, cancer mutations from causal, or ‘driver’, mutations in these data sets. Human viruses intrinsically depend on their host cell during the course of infection and can elicit pathological phenotypes similar to those arising from mutations. Here we test the hypothesis that genomic variations and tumour viruses may cause cancer through related mechanisms, by systematically examining host interactome and transcriptome network perturbations caused by DNA tumour virus proteins. The resulting integrated viral perturbation data reflects rewiring of the host cell networks, and highlights pathways, such as Notch signalling and apoptosis, that go awry in cancer. We show that systematic analyses of host targets of viral proteins can identify cancer genes with a success rate on a par with their identification through functional genomics and large-scale cataloguing of tumour mutations. Together, these complementary approaches increase the specificity of cancer gene identification. Combining systems-level studies of pathogen-encoded gene products with genomic approaches will facilitate the prioritization of cancer causing driver genes to advance the understanding of the genetic basis of human cancer.
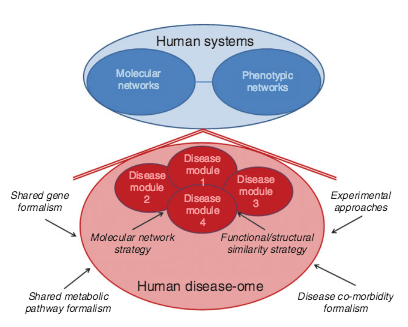
J. Loscalzo, A.-L. Barabási
Systems biology and the future of medicine
WIREs Systems Biology and Medicine 3, 619-627 (2011)
Read the abstract
Contemporary views of human disease are based on simple correlation between clinical syndromes and pathological analysis dating from the late 19th century. Although this approach to disease diagnosis, prognosis, and treatment has served the medical establishment and society well for many years, it has serious shortcomings for the modern era of the genomic medicine that stem from its reliance on reductionist principles of experimentation and analysis. Quantitative, holistic systems biology applied to human disease offers a unique approach for diagnosing established disease, defining disease predilection, and developing individualized (personalized) treatment strategies that can take full advantage of modern molecular pathobiology and the comprehensive data sets that are rapidly becoming available for populations and individuals. In this way, systems pathobiology offers the promise of redefining our approach to disease and the field of medicine.
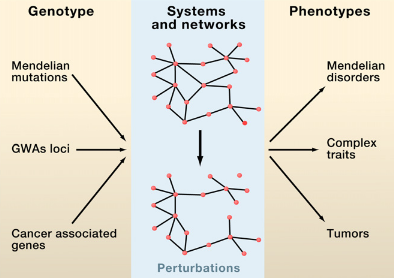
M. Vidal, M. E. Cusick, A.-L. Barabasi
Interactome Networks and Human Disease
Cell 144, 986-995 (2011)
Read the abstract
Complex biological systems and cellular networks may underlie most genotype to phenotype relationships. Here, we review basic concepts in network biology, discussing different types of interactome networks and the insights that can come from analyzing them. We elaborate on why interactome networks are important to consider in biology, how they can be mapped and integratedwith each other, what global properties are starting to emerge from interactome network models, and how these properties may relate to human disease.
J. J. Kahle, N. Gulbahce, C. A. Shaw, J. Lim, D. E. Hill, A.-L. Barabás, H. Y. Zoghbi
Comparison of an expanded ataxia interactome with patient medical records reveals a relationship between macular degeneration and ataxia
Human Molecular Genetics 20, 510-527 (2011)
Read the abstract
Spinocerebellar ataxias 6 and 7 (SCA6 and SCA7) are neurodegenerative disorders caused by expansion of CAG repeats encoding polyglutamine (polyQ) tracts in CACNA1A, the alpha1A subunit of the P/Q-type calcium channel, and ataxin-7 (ATXN7), a component of a chromatin-remodeling complex, respectively. We hypothesized that finding new protein partners for ATXN7 and CACNA1A would provide insight into the biology of their respective diseases and their relationship to other ataxia-causing proteins. We identified 118 protein interactions for CACNA1A and ATXN7 linking them to other ataxia-causing proteins and the ataxia network. To begin to understand the biological relevance of these protein interactions within the ataxia network, we used OMIM to identify diseases associated with the expanded ataxia network. We then used Medicare patient records to determine if any of these diseases co-occur with hereditary ataxia. We found that patients with ataxia are at 3.03-fold greater risk of these diseases than Medicare patients overall. One of the diseases comorbid with ataxia is macular degeneration (MD). The ataxia network is significantly (P= 7.37 × 10(-5)) enriched for proteins that interact with known MD-causing proteins, forming a MD subnetwork. We found that at least two of the proteins in the MD subnetwork have altered expression in the retina of Ataxin-7(266Q/+) mice suggesting an in vivo functional relationship with ATXN7. Together these data reveal novel protein interactions and suggest potential pathways that can contribute to the pathophysiology of ataxia, MD, and diseases comorbid with ataxia.
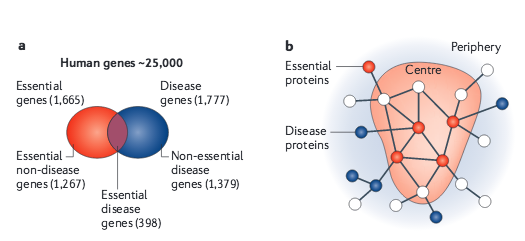
A.-L. Barabási, N. Gulbahce, J. Loscalzo
Network medicine: a network-based approach to human disease
Nature Reviews Genetics 12, 56-68 (2011)
Read the abstract
Given the functional interdependencies between the molecular components in a human cell, a disease is rarely a consequence of an abnormality in a single gene, but reflects the perturbations of the complex intracellular and intercellular network that links tissue and organ systems. The emerging tools of network medicine offer a platform to explore systematically not only the molecular complexity of a particular disease, leading to the identification of disease modules and pathways, but also the molecular relationships among apparently distinct (patho)phenotypes. Advances in this direction are essential for identifying new disease genes, for uncovering the biological significance of disease-associated mutations identified by genome-wide association studies and full-genome sequencing, and for identifying drug targets and biomarkers for complex diseases.
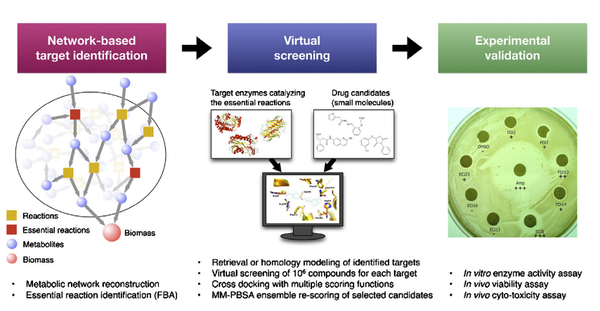
Y. Shen, L. Liu, G. Estiu, B. Isin, Y.-Y. Ahn, D.-S. Lee, A.-L. Barabásii, v. Kapatral, O. Wiest, Z. N. Oltvai
Blueprint for antimicrobial hit discovery targeting metabolic networks
Proceedings of the National Academy of Sciences of the United States of America 10.1073, 1-6 (2010)
Read the abstract
Advances in genome analysis, network biology, and computational chemistry have the potential to revolutionize drug discovery by combining system-level identification of drug targets with the atomistic modeling of small molecules capable of modulating their activity. To demonstrate the effectiveness of such a discovery pipeline, we deduced common antibiotic targets in Escherichia coli and Staphylococcus aureus by identifying shared tissue-specific or uniformly essential metabolic reactions in their metabolic networks. We then predicted through virtual screening dozens of potential inhibitors for several enzymes of these reactions and showed experimentally that a subset of these inhibited both enzyme activities in vitro and bacterial cell viability. This blueprint is applicable for any sequenced organism with high-quality metabolic reconstruction and suggests a general strategy for strain-specific antiinfective therapy.
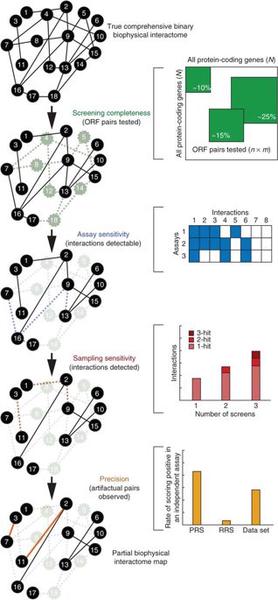
Kavitha Venkatesan, Jean-François Rual, Alexei Vazquez, Ulrich Stelzl, Irma Lemmens, Tomoko Hirozane-Kishikawa, Tong Hao, Martina Zenkner, Xiaofeng Xin, Kwang-Il Goh, Muhammed A Yildirim, Nicolas Simonis, Kathrin Heinzmann, Fana Gebreab, Julie M Sahalie, Sebiha Cevik, Christophe Simon, Anne-Sophie de Smet, Elizabeth Dann, Alex Smolyar, Arunachalam Vinayagam, Haiyuan Yu, David Szeto, Heather Borick, Amélie Dricot, Niels Klitgord, Ryan R Murray, Chenwei Lin, Maciej Lalowski, Jan Timm, Kirstin Rau, Charles Boone, Pascal Braun, Michael E Cusick, Frederick P Roth, David E Hill, Jan Tavernier, Erich E Wanker, Albert-László Barabási & Marc Vidal
An empirical framework for binary interactome mapping
Nature Methods volume 6, pages 83–90 (2009)
Read the abstract
Several attempts have been made to systematically map protein-protein interaction, or 'interactome', networks. However, it remains difficult to assess the quality and coverage of existing data sets. Here we describe a framework that uses an empirically-based approach to rigorously dissect quality parameters of currently available human interactome maps. Our results indicate that high-throughput yeast two-hybrid (HT-Y2H) interactions for human proteins are more precise than literature-curated interactions supported by a single publication, suggesting that HT-Y2H is suitable to map a significant portion of the human interactome. We estimate that the human interactome contains ∼130,000 binary interactions, most of which remain to be mapped. Similar to estimates of DNA sequence data quality and genome size early in the Human Genome Project, estimates of protein interaction data quality and interactome size are crucial to establish the magnitude of the task of comprehensive human interactome mapping and to elucidate a path toward this goal.